Article Text

Abstract
Objectives SLE is associated with endothelial cell dysfunction (ECD). Understanding how ECD leads to neutrophil infiltration into glomeruli is essential to finding therapeutic targets for SLE. The aim of this study is to determine the effect of SLE serum from patients with active disease to induce neutrophil adhesion to and chemotaxis towards glomerular endothelial cells and factors induced by serum that associate with neutrophil chemotaxis.
Methods Patients with SLE had serum collected during paired longitudinal visits with lower and higher activity. 13 patients with SLE (5 SLE, 5 SLE with hypertension (HTN) and 3 SLE lupus nephritis (LN) and HTN), and 10 healthy controls (5 with and 5 without HTN) were examined. The adhesion of neutrophils to serum-treated human renal glomerular endothelial cells (HRGECs) or chemotaxis of neutrophils towards conditioned media from serum-treated HRGECs was determined, and levels of cytokines in this conditioned medium were quantified. Pathway analysis of cytokines induced by SLE and LN serum that associated with neutrophil migration was performed.
Results HRGECs treated with SLE serum induced significantly greater neutrophil chemotaxis and adhesion compared with control serum. When examining specific cohorts, SLE HTN and LN HTN promoted greater neutrophil chemotaxis than control serum, while SLE HTN and LN HTN promoted greater chemotaxis than SLE serum. Serum from active disease visits promoted neutrophil chemotaxis and adhesion over paired inactive visits. Levels of platelet-derived growth factor-BB, interleukin (IL)-15 and IL-8 secreted by SLE serum-treated HRGECs positively correlated with neutrophil chemotaxis. Pathway analysis suggested that LN serum induced pathways important in endoplasmic reticulum and oxidative stress.
Conclusions SLE serum induces expression of mediators by HRGECs that promote neutrophil chemotaxis and adhesion, which increases during disease activity, and associates with factors common to pathways of endoplasmic reticulum and oxidative stress. These findings highlight the potential importance of serum factor-induced ECD in SLE and LN.
- lupus nephritis
- chemokines
- inflammation
- lupus erythematosus
- systemic
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE is a chronic autoimmune disease characterised by humoral autoimmunity, immune complex formation and innate and cellular immune responses. Lupus nephritis (LN) and renal failure in LN are important predictors of mortality in patients with SLE.1–5 The most aggressive form of LN is characterised by infiltration of neutrophils, fibrinoid necrosis, fibrocellular crescent formation and rupture of basement membranes.6–8 Therefore, the processes that lead to neutrophil infiltration into glomeruli are logical targets for therapy.
During homeostasis, the endothelium acts as a modulator of inflammatory responses such as cellular chemotaxis, rolling, adhesion and transmigration into tissue. When activated by inflammatory factors during pathology, the endothelium expresses chemokines and cellular adhesion molecules.9 10 Endothelial cell dysfunction (ECD), the clinical and cellular manifestation of an activated endothelium, associates with SLE disease activity.11 We have reported that endothelial nitric oxide synthase (eNOS) is essential for modulating the formation of crescentic and necrotic glomerular lesions in a murine model of LN.12 We hypothesised that factors in lupus serum would lead to glomerular ECD in a LN disease activity-dependent manner. Therefore, the first goal of the current study was to determine the effects of lupus serum on human glomerular endothelial function as assessed by neutrophil adhesion to and migration towards serum-treated human renal glomerular endothelial cells (HRGECs). The second goal was to determine potential factors and pathways leading to these cellular ECD phenotypes.
Methods
Subjects
In a prospective longitudinal cohort, patients with SLE were evaluated during standard of care visits at the Medical University of South Carolina. SLE Disease Activity Index (SLEDAI) scores13 were determined at each visit. Serum collected at each standard of care visit was stored at −80°C. Inclusion criteria from this larger cohort into this study were 1) presence of four or more American College of Rheumatology (ACR) diagnostic criteria for SLE,14 2) serum stored from two or more longitudinal, paired visits in which the SLEDAI indicating higher and lower disease activity differed by a score of ≥4 and 3) no cardiovascular disease by SLE International Collaborating Clinics Damage Index elements.15 Patients with SLE were then sorted into the following groups: 1) SLE with no nephritis (no ACR renal criteria and no visits with a urine protein/creatinine ratio >0.5 g/g) and no hypertension (HTN), 2) SLE with no nephritis but with HTN by history and 3) SLE with nephritis (active at one visit by SLEDAI renal elements or biopsy proven) and HTN. SLE demographic data are summarised in table 1. HTN was defined by the clinician as values >140/90 mm Hg for patients without renal disease and as >130/80 for patients with LN, prior to treatment with antihypertensive medications. SLE patient clinical data are summarised in table 2 and medications are summarised in table 3. Healthy controls, matched for age, sex and race, were selected if they had no cardiovascular disease by history and had a negative Connective Tissue Disease Screening Questionnaire.16 Controls were divided into those with and without HTN. Control subject clinical data are summarised in table 4. Additional healthy controls without connective tissue disease or cardiovascular risk factors/events were selected from a convenience population at the Medical University of South Carolina as donors for prospective neutrophil isolation.
Subject demographics
Clinical characteristics—lupus subjects
Medications—lupus subjects
Clinical characteristics—control subjects
HRGEC cell culture
Primary human renal glomerular endothelial cells (HRGECs) (Cat# 4000, ScienCell, Carlsbad, California, USA) were cultured according to manufacturer’s protocol. Briefly, HRGECs were cultured in fibronectin-coated flasks in complete growth media (Cat# 1001, Endothelial Cell Medium (ECM)) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement and 1% penicillin/streptomycin solution (ScienCell). Cells were incubated at 37°C in a humidified atmosphere in the presence of 5% CO2 and subcultured on reaching 90% confluency. Once confluent, cells were rinsed in Dulbecco's phosphate buffered saline (DPBS). Then 10 mL of DPBS with 1 mL 0.25% trypsin/EDTA (ScienCell) solution was added. Cells were incubated at 37 °C for 2 min or until cells were rounded. Trypsin/EDTA and cell solution was added to a conical with 5 mL FBS. Flask was incubated for another 2 min; 10 mL tyrpsin neutralization solution (TNS) solution (ScienCell) was then added to flask to collect remaining cells, and then transferred to conical with cells and FBS. Centrifuge cells at 1000 rpm for 5 min. Cells were counted and checked for viability (>90%) using Trypan Blue. Cells were then resuspended in complete growth media. HRGECs were used at passage 3–4. Based on our cell culture experience, this passage number was selected for its retention of the endothelial cell phenotype (based on CD31 staining, data not shown) and for enhanced cell viability.
Neutrophil isolation
Blood from healthy, non-hypertensive controls without cardiovascular disease was collected into K2EDTA tubes (BD) at room temperature. Less than 1 hour after blood was collected, immunomagnetic negative bead selection was performed using EasySep Neutrophil Isolation kit (StemCell Technologies, Cambridge, Massachusetts, USA) and Easy 50 magnet (StemCell Technologies) according to manufacturer’s protocol. The typical neutrophil content (CD66b+CD16+) of the final isolated fraction is 94.0%±3.7% according to manufacturer, and our lab confirmed purity by Wright-Giemsa stain. Phosphate-buffered saline (PBS) without Ca2+/Mg2+ (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 1 mM EDTA (VWR Life Science, Radnor, Pennsylvania, USA) was added to the RapidSpheres. After isolation of neutrophils, viability >90% was confirmed via Trypan Blue Solution and cells were immediately used for chemotaxis and adhesion assays. Assays were completed within 3 hours of isolation to ensure continued viability of neutrophils.
Neutrophil labelling
Neutrophils were labelled with 3 µM Calcein AM solution (Thermo Fisher Scientific) per the manufacturer’s instructions (Gibco, Thermo Fisher Scientific). Calcein AM is a cell-permeant dye used to determine cell viability, and in live cells the non-fluorescent Calcein AM is converted to a green fluorescent form by intracellular esterases. Viability of cells (>90%) is further confirmed using Trypan blue. For adhesion assays, the dyed cells were passed through a 40 µm sterile filter to remove clumps, and washed in RPMI-1640 without phenol red.
Neutrophil migration assay
HRGECs were plated at 4×104 cells per well of a 48-well fibronectin-coated plate in complete growth media at 37°C with 5% CO2. On confluence, cells were washed three times in ECM. Cells were then cultured with 10% serum (SLE or control) in ECM for 3 hours and then washed three times in ECM. Cells were serum starved overnight in ECM. The resulting conditioned media (CM) was then collected and passed through a 0.2 µm pore membrane to remove any cellular debris and frozen at −80°C for later use.
Transwell membrane chambers with 3 µm pores from a 96-well Migration Plate (Corning, Corning, New York, USA) were equilibrated in RPMI for 1 hour at 37°C. Then RPMI was removed. CM or negative (CM from serum-free treatment)/positive (10−7 M interleukin (IL)-8 (R&D Systems, Minneapolis, Minnesota, USA)) controls were added to the 96-well black-walled receiver wells (Corning); 1.5×105 neutrophils in ECM were then added to the upper chamber of each insert and incubated for 1 hour to allow for neutrophil migration toward the conditioned medium. Cells that migrated through the membrane into the CM were then labelled with Calcein AM and incubated for 1 hour at 37°C as described above. Fluorescence intensity was then measured from the bottom of the plate at 485 nm excitation, 520 nm emission using a fluorescence microplate reader (Biotek HT Synergy). A standard curve of Calcein AM-labelled neutrophils (using 1.5×105, 1.0×105, 5.0×104, 2.5×104, 1.25×104, 0.625×104 and 0 neutrophils) was created and fluorescence intensity was quantified on the microplate reader. The linear equation was then calculated and fluorescence intensity of samples could then be input into the linear equation to determine neutrophil cell number. The fluorescence intensity of migrated neutrophils in the lower chamber was measured as described.17 All values were normalised to untreated controls.
Cytokine quantification
CM was collected as indicated above for neutrophil chemotaxis migration assay. HRGECs were plated at 4×104 cells per well of a 48-well fibronectin-coated plate in complete growth media at 37°C with 5% CO2. On confluence, cells were washed three times in ECM. Cells were then cultured with 10% SLE serum (from patients with paired inactive and active disease visits, n=7) in ECM for 3 hours and then washed three times in ECM. Cells were serum starved overnight in ECM. The resulting CM was then collected and passed through a 0.2 µm pore membrane to remove any cellular debris. CM was stored at −80°C after being split into separate vials to be used for either migration assays or cytokine quantification, so that cytokine levels in CM could be directly correlated with the ability of the same CM to promote neutrophil chemotaxis. After collection, CM was stored at −80°C then shipped on dry ice to Eve Technologies for analysis. Human Cytokine Array 42-Plex with IL-18 (Eve Technologies, Alberta, Canada) and Human Supplemental Biomarker Array 10-Plex (Eve Technologies) were used to quantify cytokine concentrations. Correlation analysis with chemotaxis was then assessed. Cytokines of interest were confirmed via ELISA (R&D Systems; RayBiotech, Peachtree Corners, Georgia, USA).
Neutrophil adhesion assay
HRGECs were cultured in 48 well fibronectin-coated plates at 80 000 cells per well in complete growth media until confluent. HRGECs were washed, serum starved for 1 hour in ECM supplemented with 0.5% FBS, washed again. Treatments consisting of ECM only, 2.5% serum (lupus or control) in ECM or 100 ng/mL TNF-alpha (Thermo Fisher Scientific) (positive control) in ECM were added to the HRGECs, and cells underwent incubation for 3 hours at 37°C. HRGECs were then washed three times with RPMI-1640 (without phenol red); 6×105 Calcein AM-labelled neutrophils were added to each well of the treated HRGECs. Cells were incubated for 20 min at 37°C and washed five times with PBS (without Ca2+/Mg2+). Fluorescence intensity was quantified before the first wash and after the fifth wash to determine per cent cell adherence, with all values then normalised to untreated controls. Fluorescence intensity was measured from the top of the plate at 485 nm excitation, 520 nm emission using a fluorescence microplate reader (Biotek HT Synergy).
Statistical analysis
All samples were assayed in triplicate, and statistical analysis was performed using GraphPad Prism Software (V.8.4) or SPSS V.24.0. Depending on the distribution of the data, unpaired t-test, paired t-test or one-way analysis of variance with Tukey’s multiple comparisons were used to compare groups, while Spearman’s or Pearson’s correlation testing was performed to determine associations between variables. A generalised estimating equation was used to determine the association between cytokines and migration, accounting for two repeated measures in the same subject (during active and inactive disease). Both within-subject variables (presence of disease activity) and independent variables (cytokine levels in the conditioned medium) were modelled with neutrophil migration as the response variable.
Pathways analysis
To identify possible pathways leading to and resulting from cytokine expression, highly correlated cytokines expressed by lupus-serum-stimulated endothelial cells were identified. Reactome pathways analysis was performed.18 Overrepresentation analysis was performed to determine over representation of pathways related to the selected cytokines. Probability scores were produced and corrected for false discovery rate (FDR) using the Benjamani-Hochberg method. Results were filtered for pathways described in HomoSapiens. Only pathways with FDR p values <0.05 were selected and described.
Results
SLE serum promotes neutrophil chemotaxis to glomerular endothelial cells, which is enhanced in patients with hypertension, lupus nephritis and disease activity
Chemotaxis of inflammatory cells towards an activated endothelium is necessary for inflammation in affected tissues. In order to determine whether serum from patients with SLE impacts endothelial function, we assessed the ability of serum-treated HRGECs to induce chemotaxis of neutrophils. In these experiments, HRGECs were treated with serum, washed and serum starved, then CM from subsequent culture was collected for use in the neutrophil chemotaxis assays. CM from HRGECs treated with lupus serum induced greater neutrophil migration over that of treatment with control serum (figure 1A). When examining specific clinical phenotypes, CM from cells treated with serum from patients with SLE and HTN and SLE with LN and HTN promoted greater neutrophil chemotaxis than treatment with control serum or SLE without HTN serum (figure 1B). The effect of serum from paired inactive and active disease visits was then compared with determine whether SLE activity-related factors promote neutrophil chemotaxis. CM from HRGECs treated with serum from patients during active disease visits (mean=2.56, SD=0.96) induced greater chemotaxis than that from paired inactive disease visits (mean=2.26, SD=0.91) (t=2.46, df=12, p=0.03) (figure 1C).
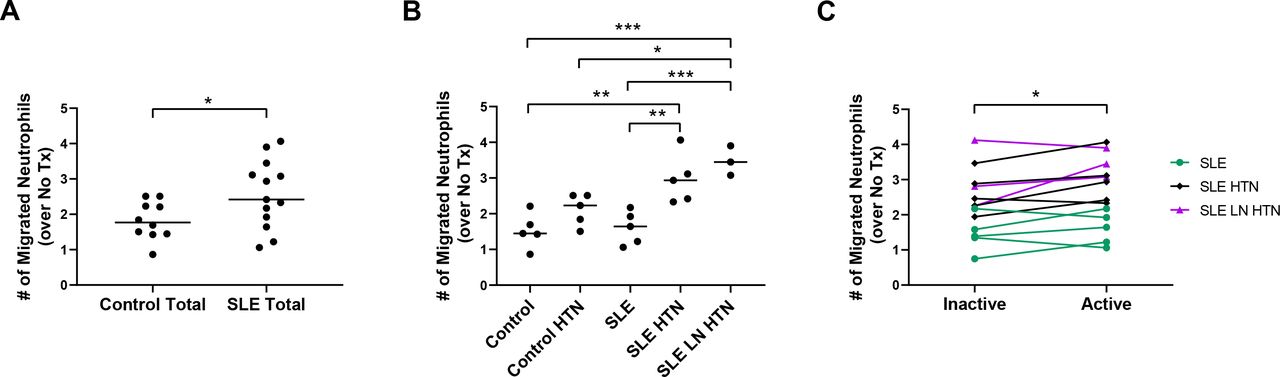
SLE serum promotes neutrophil chemotaxis, which is enhanced with hypertension (HTN), lupus nephritis (LN) and disease activity. (A) Neutrophil migration towards conditioned media from HRGECs treated with control (control, control HTN) or SLE with active disease (SLE, SLE HTN, SLE LN HTN) serum or B) migration of neutrophils to conditioned media from HRGECs treated with serum from donor patient groups: control, control with HTN, SLE with active disease, SLE with HTN with active disease and SLE with LN and HTN with active disease. (C) Migration of neutrophils to conditioned media from HRGECs treated with serum from patients with SLE during paired inactive and active disease visits. Lines represent median values. Results are representative of three experiments, with each experiment using a different healthy neutrophil donor. Statistical analysis was by two-tailed unpaired t-test (A), one-way analysis of variance with Tukey’s multiple comparisons (B) and two-tailed paired t-test (C). ***P<0.001, **p<0.01, *p<0.05.
Cytokines secreted by SLE serum-exposed HRGECs correlate with neutrophil chemotaxis
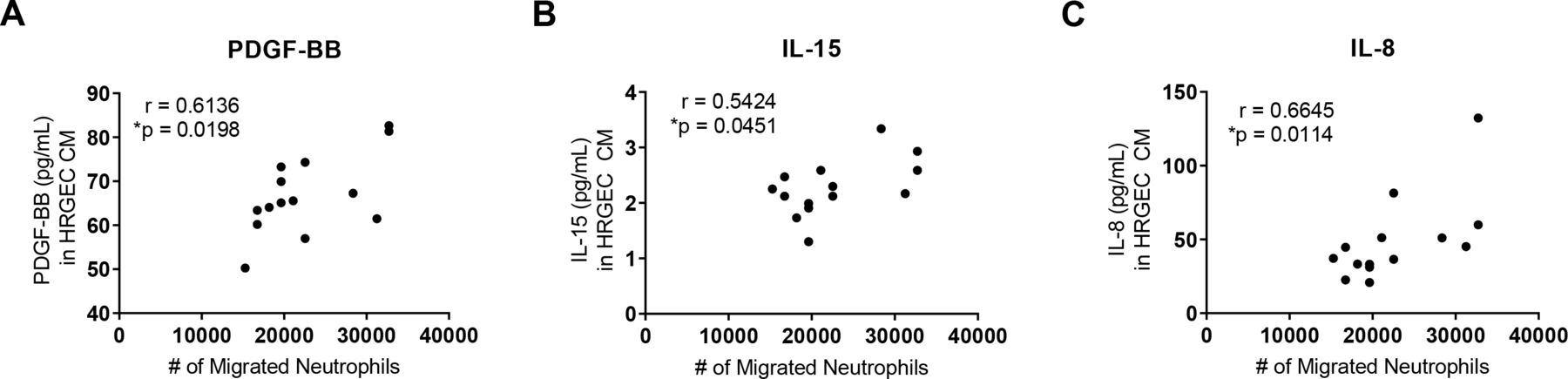
To explore which factors are released by serum-activated endothelial cells that may contribute to neutrophil migration, we analysed the concentration of candidate factors in the CM by a multiplex array. We correlated the levels of those factors with the migration of neutrophils towards the same CM. Levels of platelet-derived growth factor-BB (PDGF-BB) (figure 2A), IL-15 (figure 2B) and IL-8 (figure 2C) all positively correlated with neutrophil migration. In figure 1, we found that disease activity, HTN and renal involvement led to differences in neutrophil chemotaxis; however, we did not find differences between these groups in the cytokine profiles (online supplemental figures 1-3).
Supplemental material
Supplemental material
Supplemental material
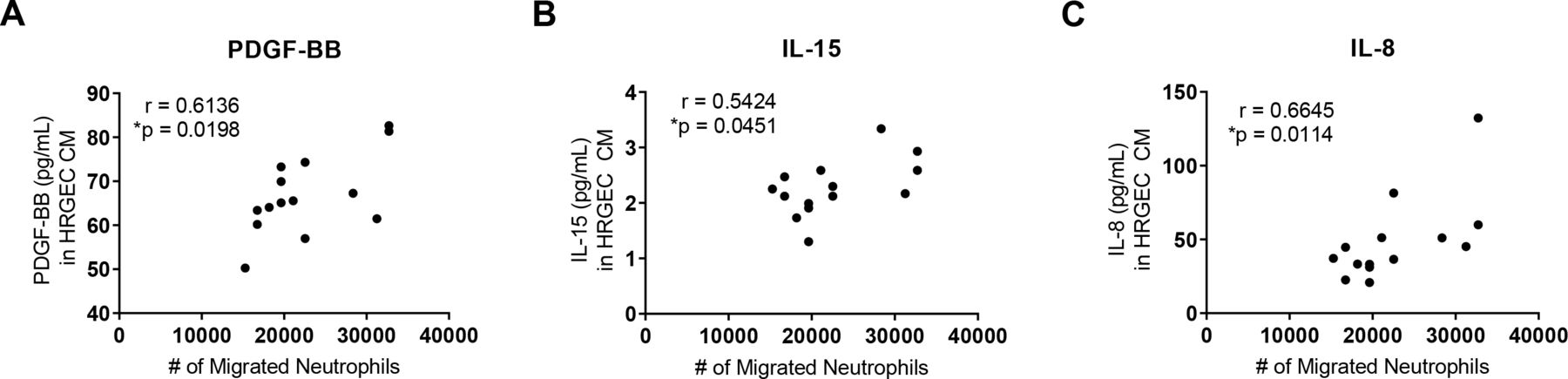
Cytokines secreted by SLE serum-exposed human renal glomerular endothelial cells (HRGECs) correlate with neutrophil chemotaxis. Cytokine array of conditioned medium from HRGECs treated with SLE serum collected during inactive and active disease (n=7 for paired inactive and active disease visits) revealed chemotactic factors (A) platelet-derived growth factor-BB (PDGF-BB), (B) interleukin (IL)-15 and (C) IL-8 that correlated with neutrophil migration to the same conditioned medium. Results are representative of two experiments. Statistical analysis was by Spearman’s correlation. *P<0.05.
Pathways common to highly correlated induced factors are important in oxidative stress and endoplasmic reticulum stress
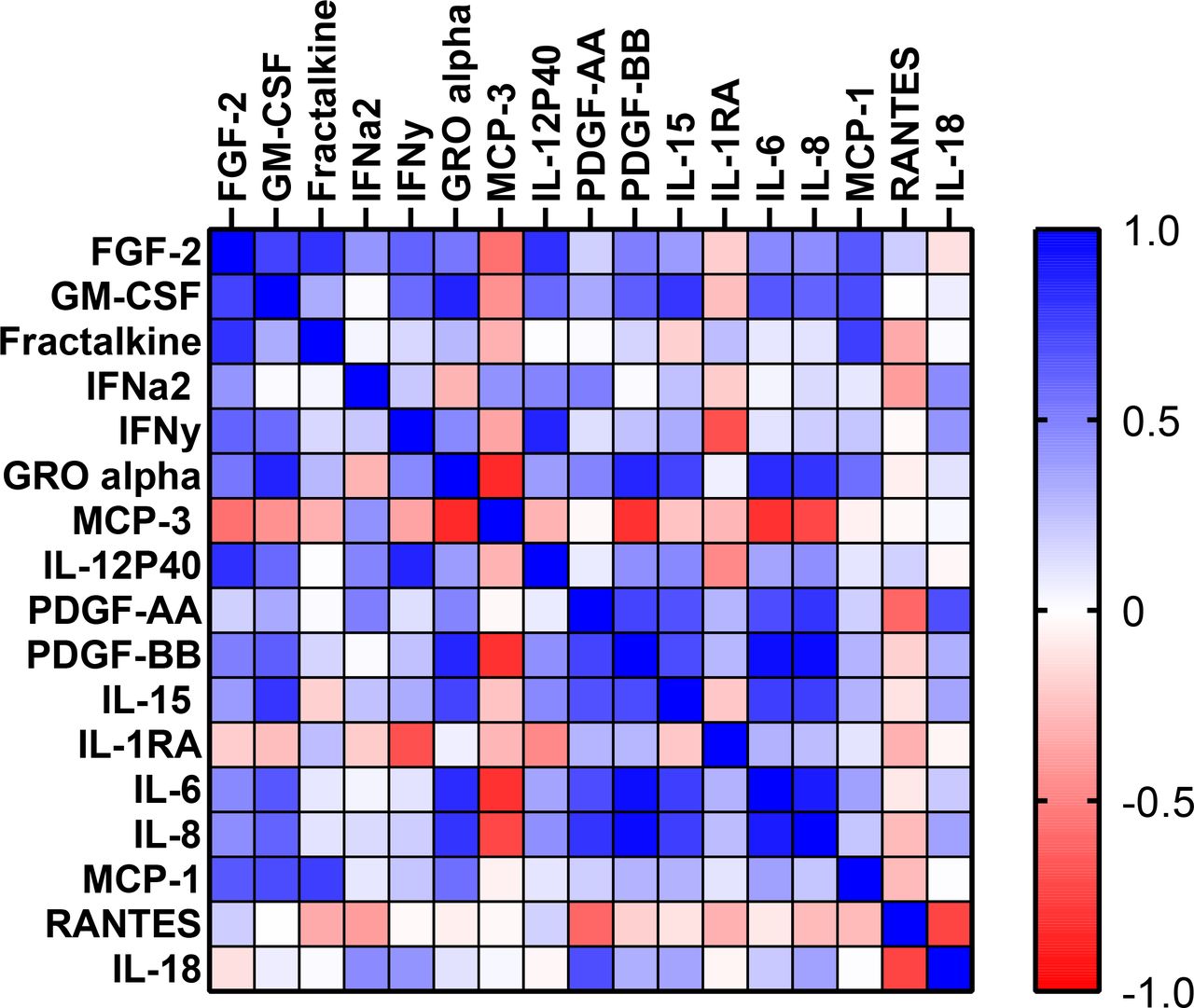
To suggest common pathways induced by SLE serum during disease activity, we determined which secreted factors in the CM from serum-treated endothelial cells were highly correlated with each other (figure 3). PDGF-BB positively correlated with IL-8 and IL-6, with IL-8 and IL-6 positively correlating with each other. Growth-regulated oncogene alpha (GRO-alpha or CXCL1) positively correlated with granulocyte macrophage colony stimulating factor, PDGF-BB and IL-6 (table 5). To understand further the functional relationships of some of these factors, we used the Reactome Knowledgebase to ascertain which pathways related to IL-8, PDGF-BB and IL-15 may be involved in neutrophil migration.19 The most significant pathways were intuitive and included: immune system, cytokine signalling, IL-2 family signalling (including IL-15 signalling), signalling by PDGF, non-integration membrane-ECM interactions, chemokine receptors and downstream signal transduction. However, a few were of interest that were less intuitive, including: activating transcription factor 4 (ATF4), protein kinase R-like endoplasmic reticulum kinase (PERK) regulation of gene expression and IL-10 signalling (see online supplemental data).
Supplemental material
Correlation between cytokines expressed by HRGECs stimulated with lupus serum

Correlation matrix of factors in conditioned medium from SLE from serum-treated human renal glomerular endothelial cells (HRGECs). The correlation between levels of all cytokines from conditioned medium in the experiment from figure 2 was determined and displayed as a correlation matrix. Blue boxes reflect positive correlations, and the darkness of the box reflects the r value. Similarly, red indicates a negative correlation, and darkness of the box reflects the r value calculated by Spearman’s correlation. GM-CSF, granulocyte macrophage colony stimulating factor
SLE serum promotes neutrophil adhesion to HRGECs
One of the hallmarks of endothelial dysfunction is adhesion of circulating neutrophils to the endothelium. The impact of lupus serum on endothelial dysfunction was determined by examining neutrophil adhesion to washed HRGECs after treatment with either control or lupus serum. HRGECs treated with lupus serum increased neutrophil adhesion over that of control serum (figure 4A). While trends were present, no significant difference was seen between control, control HTN, SLE, SLE HTN and SLE LN HTN serum treatment (figure 4B). Endothelial cells treated with serum from patients during active disease visits (mean=1.97, SD=0.69) induced greater neutrophil adhesion than those treated with serum from the same patients during paired visits with inactive disease (mean=1.543, SD=0.56) (t=3.48, df=10, p=0.006) (figure 4C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
SLE serum promotes neutrophil adhesion, which is enhanced with disease activity. (A) Neutrophil adhesion to human renal glomerular endothelial cells (HRGECs) treated with control (control, control hypertension (HTN)) or SLE with active disease (SLE, SLE HTN, SLE lupus nephritis (LN) HTN) serum or (B) adhesion of neutrophils to HRGECs treated with serum from donor patient groups: control, control with HTN, SLE with active disease, SLE with HTN with active disease and SLE with LN and HTN with active disease. (C) Adhesion of neutrophils to HRGECs treated with serum from patients with SLE during paired inactive and active disease visits. Lines represent median values. Results are representative of three experiments, with each experiment using a different healthy neutrophil donor. Statistical analysis was by two-tailed unpaired t-test (A), one-way analysis of variance with Tukey’s multiple comparisons (B) and two-tailed paired t-test (C). ***P<0.001, **p<0.01.
Discussion
This study demonstrated that factors in SLE serum can induce an inflammatory response that enhances neutrophil migration and adhesion in human glomerular endothelial cells. Neutrophil migration was shown to increase with SLE HTN and LN HTN compared with control, and also increased with disease activity, while neutrophil adhesion increased with disease activity. Chemokine production (IL-8, IL-15 and PDGF-BB) by SLE and LN serum-stimulated HRGECs associated with migration of neutrophils to the same medium. Taken together, these findings suggest that glomerular endothelial cells play an active role in the inflammatory response in lupus and LN, and that unique factors in the serum induce this response.
A heterogeneity of factors in SLE serum may lead to this response. Several reports suggest receptor-mediated and redox signalling mechanisms lead to endothelial dysfunction.20–26 Our laboratory has published that eNOS is uncoupled with addition of lupus serum.17 This uncoupling leads to production of reactive oxygen species in an animal model of LN and in LN glomerular tissue.12 27 Therefore, reduction oxidation (redox) signalling may be a common intracellular mechanism in endothelial cell activation. Factors contained in microparticles and immune complexes also lead to endothelial cell activation.26 Isolated chemokines known to be associated with LN can, in combination, induce expression of inflammatory cytokines and growth factors in immortalised glomerular endothelial cells.28
HTN was associated with a serum-induced chemotaxis of neutrophils to HRGECs. HTN itself leads to endothelial dysfunction. Angiotensin signalling via the angiotensin II receptor induces NADPH oxidase-dependent reactive intermediate production in endothelial cells and reduces the modulating effect of eNOS29 by an uncoupling of the eNOS homodimer.30 These combined findings support the hypothesis that endothelial dysfunction induced by HTN and lupus are either synergistic or share common mechanisms.
This study suggests several mechanisms for serum-induced neutrophil migration that are biologically plausible in SLE. PDGF-BB is chemotactic for neutrophils and monocytes and may act in to induce vascular injury in LN.31 While the literature on expression of functional PDGF receptors on neutrophils is conflicting,32 endothelial cells express PDGFRB, which signals through phosphatidylinositol 3-kinase activation.33 In turn, PDGF-BB can act in an autocrine fashion to stimulate endothelial production of CCL2/monocyte chemotactic protein-1 (MCP-1).34 IL-8, on the other hand, is chemotactic to neutrophils through neutrophil expression of CCR1 and CCR2.35 IL-8 also induces neutrophil oxidative burst and extracellular traps formation.36 Urine levels of this factor are associated with lack of response to therapy in human LN,37 and a polymorphism of IL-8 is associated with poor outcome in African-Americans.38 IL-15 acts as a chemokine to several cell types including neutrophils, which express the IL-15Rα chain.39 Levels of IL-15 are elevated in humans with lupus.40
This study suggests that LN serum may activate pathways known to be involved in oxidative stress and endoplasmic reticulum (ER) stress. During ER redox imbalance, ER stress can induce dissociation of HSP90 from PERK, which decreases HSP90 stability and causes the dissociation of eNOS from HSP90, leading to eNOS uncoupling.41 Furthermore, PERK can phosphorylate and activate ATF4 to induce expression of anti-oxidative genes.42–44 ATF4 is activated in ER stress in endothelial cells and in turn induces expression of chemokines such as IL-8 and MCP-145 46 in response to oxidised lipids. Thus, the PERK/ATF4 axis plays an important role in cellular stress conditions, including oxidative stress which greatly impacts the functionality of the endothelium. Lastly, IL-10 is known to inhibit nitric oxide production, inhibit endothelial cell differentiation and repair and may enhance the negative effect of interferon-alpha on endothelial function.47
Of note, low-density lipoprotein (LDL) was higher in some patients with active disease. While these differences did not achieve statistical significance (p=0.37 for total cholesterol and p=0.32 for LDL), they are consistent with reports by Durcan et al in which LDL levels correlated with disease activity.48 Because we did not measure oxidised LDL, we cannot determine if LDL may have induced endothelial dysfunction through oxidised LDL receptors.
This study has several limitations. First, patients in this study were prescribed a variety of medications (table 3). This concern is tempered by the fact that, in a separate analysis, we found no difference in either neutrophil migration or adhesion when examining dosing effects of Plaquenil, Prednisone, Lisinopril or CellCept (online supplemental figures 4 and 5). However, when examining use of immunomodulatory medications (as a group) or ACE inhibitors/angiotensin receptor blockers, we found increased neutrophil migration with use of both medication groups and increased neutrophil adhesion for immunomodulatory medications (online supplemental figures 6 and 7). This is the opposite effect of what we anticipated and may indicate a bias from indication. It also suggests factors in serum that are important for adhesion and migration of neutrophils to the endothelium may not be controlled by these medications. Second, this was a retrospective study. However, the samples were collected in a prospective, longitudinal manner in a well-characterised cohort of patients. In addition, there was an association between migration and adhesion and disease activity in paired samples from the same patients. This reduces some of the variability seen within a population in cross-sectional studies and reveals significant differences even in a limited number of samples in each lupus phenotype. Third, the chemokines induced by LN serum were analysed as candidates based on the literature, and it is possible that pathogenic chemotactic factors were not included in the analysis. Fourth, interassay variability was introduced using multiple healthy donors of neutrophils. However, the migration and adhesion rank order remained similar across experiments (data not shown). Fifth, the choice of 2.5% serum does not reflect the intravascular environment seen in patients. This concentration was chosen, as it was the highest concentration at which lupus serum did not induce endothelial cell damage in concentration titration experiments (data not shown). Finally, we did not compare the effects of serum from patients with other renal diseases such as diabetic renal disease. While we chose to focus on SLE in this study, diabetic and hypertensive renal disease are also known to be associated with endothelial dysfunction that improves with therapy targeting eNOS dysfunction.49–51
Supplemental material
Supplemental material
Supplemental material
Supplemental material
This study established that factors in the serum of patients with lupus and LN induce an endothelial cell inflammatory phenotype in renal glomerular endothelial cells, which is manifested by neutrophil adhesion and migration. It also demonstrates the effect of disease activity on this phenotype using paired visit serum samples from individual patients during inactive and active disease states. This study demonstrates associations between migration of neutrophils and serum-induced chemokine production in glomerular endothelial cells. Common pathways associated with these factors are important in ER stress, oxidative stress and uncoupling of eNOS. The findings from this study reinforce the importance of glomerular endothelial cells in trafficking neutrophils to sites of inflammation through generation of pathogenic chemotactic factors. They further provide the rationale for studying redox-induced pathways and ER stress as mechanisms for glomerular inflammation in LN.
Acknowledgments
The authors would like to thank Dr John Zhang for his helpful scientific input, SCTR Institute and Amy Chamberlain and Claire Tyson for coordination/collection of control blood specimens and the MUSC Rheumatology Biorepository team for collection and distribution of SLE serum and lupus phenotype data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JO conceived the idea and directed the project. DR and MM designed and performed the experiments, derived the models and analysed the data. DR and JO wrote the manuscript in consultation with MM.
Funding This work was supported by funding from the Department of Veterans Affairs (CXC001248-01A2 (Oates)). Data and materials as well as personnel time were made possible by the following funding: K08AR002193 (Oates)), NIH NIAMS P60AR062755 and NIAMS P30AR072582 (Oates).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the Medical University of South Carolina Institutional Review Board protocols: Pro00052903 Pro00056002. All human subject research was in compliance with the Helsinki Declaration. Subjects were enrolled after written informed consent was provided.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.