Article Text

Abstract
Objectives To evaluate the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of single-dose and multiple-dose administration of AMG 557, a human anti-inducible T cell co-stimulator ligand (ICOSL) monoclonal antibody, in subjects with systemic lupus erythematosus (SLE).
Methods Patients with mild, stable SLE (n=112) were enrolled in two clinical trials to evaluate the effects of single (1.8–210 mg subcutaneous or 18 mg intravenous) and multiple (6 –210 mg subcutaneous every other week (Q2W)×7) doses of AMG 557. Subjects received two 1 mg intradermal injections 28 days apart of keyhole limpet haemocyanin (KLH), a neoantigen, to assess PD effects of AMG 557. Safety, PK, target occupancy, anti-KLH antibody responses, lymphocyte subset analyses and SLE-associated biomarkers and clinical outcomes were assessed.
Results AMG 557 demonstrated an acceptable safety profile. The PK properties were consistent with an antibody directed against a cell surface target, with non-linear PK observed at lower concentrations and linear PK at higher concentrations. Target occupancy by AMG 557 was dose dependent and reversible, and maximal occupancy was achieved in the setting of this trial. Anti-AMG 557 antibodies were observed, but none were neutralising and without impact on drug levels. A significant reduction in the anti-KLH IgG response was observed with AMG 557 administration without discernible changes in the anti-KLH IgM response or on the overall IgG levels. No discernible changes were seen in lymphocyte subsets or in SLE-related biomarkers and clinical measures.
Conclusions The selective reduction in anti-KLH IgG demonstrates a PD effect of AMG 557 in subjects with SLE consistent with the biology of the ICOS pathway and supports further studies of AMG 557 as a potential therapeutic for autoimmune diseases.
Trial registration numbers NCT02391259 and NCT00774943.
- B cells
- Inflammation
- Systemic Lupus Erythematosus
- T Cells
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Lymphocyte co-stimulation plays a central role in immunology, inflammation and immunotherapy.1–4 The inducible T cell co-stimulator (ICOS) is expressed on T cells following peptide:MHC engagement with CD28 co-stimulation. The interaction of ICOS with its sole ligand the Inducible T-cell co-stimulatory ligand (ICOSL; also known as B7-related protein-1 or ICOSL) triggers key activities of T cells including cytokine production and differentiation into the T follicular helper (Tfh) cell lineage over effector lineages.5 ,6 Tfh cells help initiate and maintain the germinal centre (GC) reaction,7–9 dynamically selecting high-affinity, antibody-producing B cells for clonal expansion.10 Phenotypic characterisations of patients with an ICOS null genotype demonstrate significant reductions in circulating memory Tfh, class-switched memory B cells and serum IgG (while maintaining normal to elevated IgM levels),11–14 consistent with mouse studies of ICOS deficiency.15–17 ICOS is also critical for T effector memory differentiation and contributes to the development of Th1, Th2 and Th17 cell lineages.14 ,16 ,18–23 Dysregulation of the pathway through a point mutation in RC3H1 (roquin 1), a repressor of ICOS, leads to the overexpression of ICOS and the development of a lupus-like systemic autoimmune disease in mice.24–26 Pharmacological blockade and use of knockout rodents in animal models of autoimmune diseases have demonstrated the potential for therapeutic intervention of this pathway in human autoimmune diseases such as systemic lupus erythematosus (SLE), asthma and rheumatoid arthritis (RA).27–30
SLE is a multisystem autoimmune disease of unknown cause with diverse clinical manifestations that disproportionately affects minorities and women of childbearing potential. The presence of class-switched IgG autoantibodies is a hallmark of this disease, implicating immune dysregulation as a driving force for disease pathogenesis.31–34 ICOS is elevated on peripheral T cells in patients with SLE, RA and primary Sjögren's syndrome (pSS).35–38 In patients with SLE, enhanced ICOS expression is a stable phenotype and the magnitude of ICOS expression correlates with anti-dsDNA titre.36 ICOS+ T cells co-localise and form close associations with B cells in SLE nephritis lesions35 ,39 and Tfh (CD3+Bcl6+) are enriched in pSS minor salivary gland ectopic GCs,40 suggesting both central and peripheral dysregulation.
AMG 557 is a fully human IgG2 monoclonal antibody that binds to ICOSL and prevents functional interaction with ICOS on activated T cells. It does not cross-react with other B7 co-stimulatory molecule family members. In this report, we describe the clinical and immunological impact of single-dose and multiple-dose administration of AMG 557 in subjects with stable, mild SLE, translating the findings in mouse models into the clinic. AMG 557 demonstrated that therapeutic blockade of ICOSL leads to the selective inhibition of the antigen-specific IgG antibody response in a dose-dependent manner in keeping with the known biology of this co-stimulatory pathway.
Materials and methods
Study design
The study designs and dose-escalation schedule are summarised in figure 1. The studies were carried out in accordance with the ethical principles set forth in the Declaration of Helsinki and the International Conference on Harmonization E6 Guidance for Good Clinical Practice. The sites and ethics committees that granted approval for these studies are listed in online supplementary appendix A.
Supplementary appendix

Study schemas. Upper panel: single-ascending dose study. Subjects with mild, stable systemic lupus erythematosus (SLE) were randomised to receive placebo or AMG 557 at 1.8, 6, 18, 60, 140 and 210 mg subcutaneously (SC) or 18 mg intravenously (IV) in seven sequential rising-dose cohorts. Subjects were immunised with both a prime and boost consisting of a 1 mg intradermal injection of keyhole limpet haemocyanin (KLH) on days 2 and 29 (for 60 mg SC cohort), days 8 and 36 (for 140 and 210 mg SC cohorts), and days 15 and 43. Lower panel: multiple-ascending dose study. Subjects with mild, stable SLE were randomised to receive placebo or AMG 557 at 6, 18, 30, 45, 70, 140 or 210 mg subcutaneously in seven sequential rising-dose cohorts. Subjects were administered AMG 557 or placebo on days 1, 15, 29, 43, 57, 71 and 85. Subjects were immunised with KLH on days 57 and 85. EOS, end of study.
Both studies were registered on clinicaltrials.gov. All subjects involved in both studies signed the study-specific informed consent forms indicating their consent to participate in the study. For both studies, eligible subjects were men and women 18–70 years of age with a diagnosis of SLE as defined by the most recent American College of Rheumatology criteria including a positive antinuclear antibodies (ANAs) test at screening or documented positive ANA in the past. The phase Ia single-ascending dose (SAD) study was a randomised, double-blind, placebo-controlled study in patients with SLE with mild, stable disease (ie, patients without anticipated changes to therapy or severe manifestations such as active systemic vasculitis and who were on ≤10 mg/day of prednisone or equivalent). Fifty-six subjects with mild stable SLE were randomised to receive placebo or AMG 557 at 1.8, 6, 18, 60, 140 and 210 mg subcutaneously or 18 mg intravenously in seven sequential rising-dose cohorts. Subjects were immunised with both a prime and boost consisting of a 1 mg intradermal injection of keyhole limpet haemocyanin (KLH) (Vacmune, Biosyn, Carlsbad, California, USA) on days 2 and 29 (for the 60 mg subcutaneous cohort), and days 8 and 36 (for the 140 and 210 mg subcutaneous cohorts). Two subjects in the 210 mg cohort received KLH injections on days 15 and 43.
The phase Ib multiple-ascending dose (MAD) study was a randomised, double-blind, placebo-controlled study in patients with SLE with mild, stable disease. Fifty-six subjects with SLE were randomised to receive placebo or AMG 557 at 6, 18, 30, 45, 70, 140 or 210 mg subcutaneously in seven sequential rising-dose cohorts. Subjects were administered AMG 557 or placebo on days 1, 15, 29, 43, 57, 71 and 85. Subjects were immunised with both a prime and boost consisting of a 1 mg intradermal injection of KLH on days 57 and 85. In the MAD study, although all 56 subjects were planned per protocol to be immunised with KLH, 51 subjects were immunised and 5 did not follow the protocol and were not immunised.
Detection of AMG 557 in serum
AMG 557 serum levels were measured using a validated, quantitative, ELISA developed at Amgen, Seattle, Washington, USA. Biotin rabbit anti-AMG 557 was used as capture antibody, and mouse mAb anti-AMG 557 and followed by horseradish peroxidase (HRP)-conjugated goat antimouse Fc were added as detection antibodies. One-component 3,3′,5,5′-tetramethylbenzidine substrate solution was added to react with peroxide. A colorimetric signal that was proportional to the amount of AMG 557 bound by the capture reagent was developed and measured. Standards and quality controls were prepared by spiking AMG 557 into 100% normal human serum pool. The lower limit of quantification was 15 ng/mL.
Detection of anti-AMG 557 antibodies in serum
A bridging assay was used to detect anti-AMG 557 binding antibodies using AMG 557 to capture serum antidrug antibodies and—biotin labelled—AMG 557 (produced at Amgen, Thousand Oaks, California, USA), followed by streptavidin-HRP to detect the bridged binding anti-AMG 557 antibodies. Samples that tested positive in the binding assay were measured for neutralising antibodies. This assay tested serum sample for restoring of interruption caused by AMG 557 of ICOS:ICOSL binding in vitro using recombinant ICOS and Fc-fusion-ICOSL (produced at Amgen) in a receptor-binding assay.
Detection of target occupancy on circulating B cells
ICOSL target occupancy was assessed using a validated, whole blood flow cytometric assay that used two phycoerythrin (PE)-conjugated monoclonal antibodies produced at Amgen: one that competed with AMG 557 (ROA-1, to count ‘free’ ICOSL) and one that did not compete (ROA-2, to count ‘total’ ICOSL). The two antibodies were produced at Amgen. Free and total ICOSL median fluorescence intensity (MFI) values were converted to Molecules of Equivalent Soluble Fluorochrome using a fluorescent bead standard curve (Bangs Laboratories, Fishers, Indiana, USA).
The per cent target occupancy for each subject's sample was calculated as [1 − (the ratio of free to total ICOSL) divided by (the subject's baseline ratio of free to total ICOSL)] times 100%. Day 1 predose samples were used as baseline. Mean target occupancy for each dose level and study visit was estimated using a mixed effects linear model including factors for dose, visit and the interaction between dose and visit. Subject was included as a random factor. The intervals shown are the group means±95% CIs around the mean.
Detection of anti-KLH IgM and IgG antibodies in serum
KLH-specific IgM and IgG levels in the serum were measured using a validated, semiquantitative flow cytometric bead array as described by Ferbas et al,41 modified to include an anti-KLH IgG standard curve. Briefly, serum was incubated with a mixture of fluorescently labelled beads each individually conjugated to KLH, tetanus toxoid (TT), sperm whale myoglobin (SWM), IgG and IgM (manufactured by Becton Dickinson, La Jolla, California, USA). The beads were washed and incubated with a PE-mouse (Fab) antihuman IgG or a PE-mouse (Fab) antihuman IgM (both provided by Becton Dickinson) and detected by flow cytometry (FACSCalibur, Becton Dickenson, San Jose, California, USA). For the anti-KLH IgG assay, the MFI of the anti-KLH bead was converted to ng/mL using an anti-KLH standard curve consisting of monoclonal human anti-KLH IgG1, IgG2, IgG3 and IgG4 (produced at Amgen). Anti-KLH IgM data are shown as MFI as a recombinant anti-KLH IgM was not available. The beads conjugated to SWM, TT, IgG and IgM served as assay positive and negative controls.
Detection of anti-TT IgG and total IgG antibodies in serum
Serum tetanus antitoxoid levels were measured by a quantitative ELISA (Focus Diagnostics, Cypress, California, USA). Results are reported in IU/mL. Serum IgG levels were measured by a quantitative fluorescent enzyme immunoassay (Focus Diagnostics). Results are reported in mg/dL. Serum samples were analysed on days –1, 85, 169 and 253 (end of study). Three (of 56) subjects were excluded from this analysis because they only received 2–3 doses of AMG 557.
Results
Baseline characteristics and clinical findings
This is a combined report of two clinical trials that enrolled subjects with mild, stable SLE (figure 1 and table 1). Fifty-six subjects were enrolled in the phase I, SAD (1.8–210 mg subcutaneous and 18 mg intravenous) trial. Most subjects in this study were women, and the majority were white. The mean age was 44.8 years in the AMG 557 group and 48.5 years in the placebo group. ANAs were positive at baseline for 83% in the AMG 557 group and 85% in the placebo group. The overall percentages of patients with anti-dsDNA antibodies or hypocomplementemia were low. The mean British Isles Lupus Assessment Group index (BILAG) baseline scores and the Systemic Lupus Erythematosus Disease Activity Index SELENA Modification (SELENA-SLEDAI) baseline scores were generally low, reflecting the protocol-specified population. Fifty-six subjects were enrolled in the phase Ib, MAD trial (6 –210 mg subcutaneous (Q2W×7)). Most subjects were women and white with a mean age of 48.8 years in the AMG 557 group and 48.2 years in the placebo group. A relatively low percentage of subjects had positive ANA at baseline (62% in the AMG 557 group and 64% in the placebo group), although a documented history of positive ANA was a requirement of study entry. Overall, there were low numbers of subjects with anti-dsDNA antibodies or hypocomplementemia.
Subject demographics and baseline characteristics
Single-dose and multiple-dose administration of AMG 557 was generally well tolerated and demonstrated an acceptable safety profile in subjects with mild stable SLE. The adverse events were classified by the investigators as non-KLH-related or KLH-related. In both studies, the incidence of non-KLH-related adverse events did not appear to increase with increasing AMG 557 dose and was similar between the AMG 557 and placebo-treated groups. No deaths were reported. Serious adverse events were not reported in the AMG 557 group in the SAD study, but were reported for five subjects (12%) who received AMG 557 and two subjects (14%) who received placebo in the MAD study. In the MAD study, one subject in the 210 mg dose cohort experienced trilobar pneumonia and had a serious adverse event of respiratory failure 95 days after the final dose of AMG 557 (study day 181) that resulted in the subject's withdrawal from study and was considered by the investigator and Amgen to be related to AMG 557. The incidence of KLH-related adverse events (consisting mostly of injection-site reactions) was similar between the AMG 557 and placebo groups in both studies and was graded as mild or moderate in severity.
Consistent changes in disease activity assessed with BILAG and SELENA-SLEDAI, anti-dsDNA and complement levels were not observed. Measurable change in disease activity was not anticipated in these subjects with stable, mild disease enrolled in this study.
Pharmacokinetics and immunogenicity
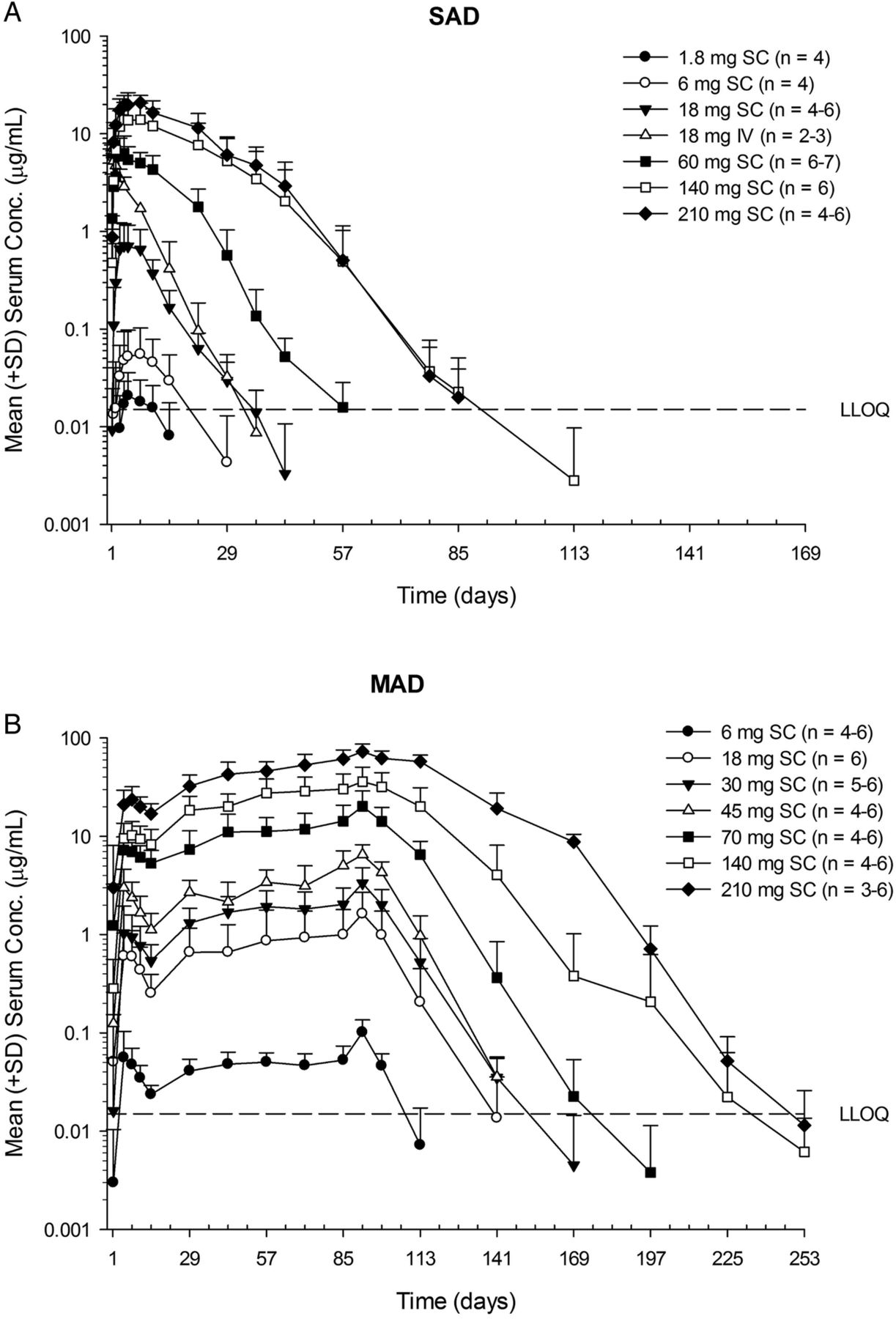
Serum AMG 557 exposure (Cmax and area under the curve (AUC)) increased greater than dose-proportionally at lower doses after either single or multiple subcutaneous doses (figure 2 and table 2). However, an approximately dose-proportional increase in exposure was observed at higher doses (≥140 mg for SAD;≥70 mg for MAD). Maximum concentration of AMG 557 was reached in 3–7 days (tmax) after subcutaneous administration (table 2). Bioavailability after subcutaneous dosing was estimated to be 57%. Moderate accumulation was observed at steady state (seventh dose), with median accumulation ratios being approximately 2–4. A total of 6% (N=2/36, SAD) and 24% (N=10/42, MAD) of AMG 557-treated subjects tested positive for postbaseline anti-AMG 557 binding antibodies, although anti-AMG 557 antibodies were present in 9% (5/56) of subjects at baseline in the MAD study, which may be due to cross-reactivity in the assay. The specificity of the anti-AMG 557 assay was determined by addition of 100 µg/mL of excess unlabelled drug to the samples that tested positive in the assay. If the addition of the excess drug quenched the positive signal in the assay, they are considered specific to the drug. There was no apparent impact of anti-AMG 557 antibodies on serum AMG 557 concentrations. No subjects tested positive for neutralising anti-AMG 557 antibodies.
Descriptive statistics for pharmacokinetic parameter estimates of AMG 557 after subcutaneous or intravenous administration of AMG 557 to subjects with systemic lupus erythematosus

Mean (SD) AMG 557 serum concentration–time profiles following single-ascending dose (SAD) (A) and multiple-ascending dose (MAD) (B) administration AMG 557 in subjects with systemic lupus erythematosus. The dose levels are indicated in the legend, with the number of values per symbol in parentheses. The lower limit of quantitation (LLOQ) of 15 ng/mL is indicated as a dashed horizontal line on each graph. IV, intravenous; SC, subcutaneous.
Target occupancy
Relative target occupancy was assessed on B cells in the peripheral blood using a semiquantitative flow cytometric assay in both studies. Overall, the degree of ICOSL target occupancy by AMG 557 was found to be dose-related and reversible, sustaining maximal levels in the higher dose groups in the MAD study (figure 3A). Similar results were observed with the SAD study. There was a dose-related increase in total ICOSL levels on peripheral B cells and a dose-related decrease in free ICOSL on peripheral B cells (data not shown), consistent with preclinical studies of a mouse surrogate anti-ICOSL antibody.42 The relative RO in figure 3A was calculated using baseline-free and total ICOSL as a comparator. The relative RO was at times ‘negative’ due to a fall in total ICOSL levels and a rise in free ICOSL levels after the last dose beyond measured baseline levels, resulting in a negative number using the equation as described in ‘Materials and methods’. This is a consequence of the fluctuating levels of ICOSL and the variability of the assay.
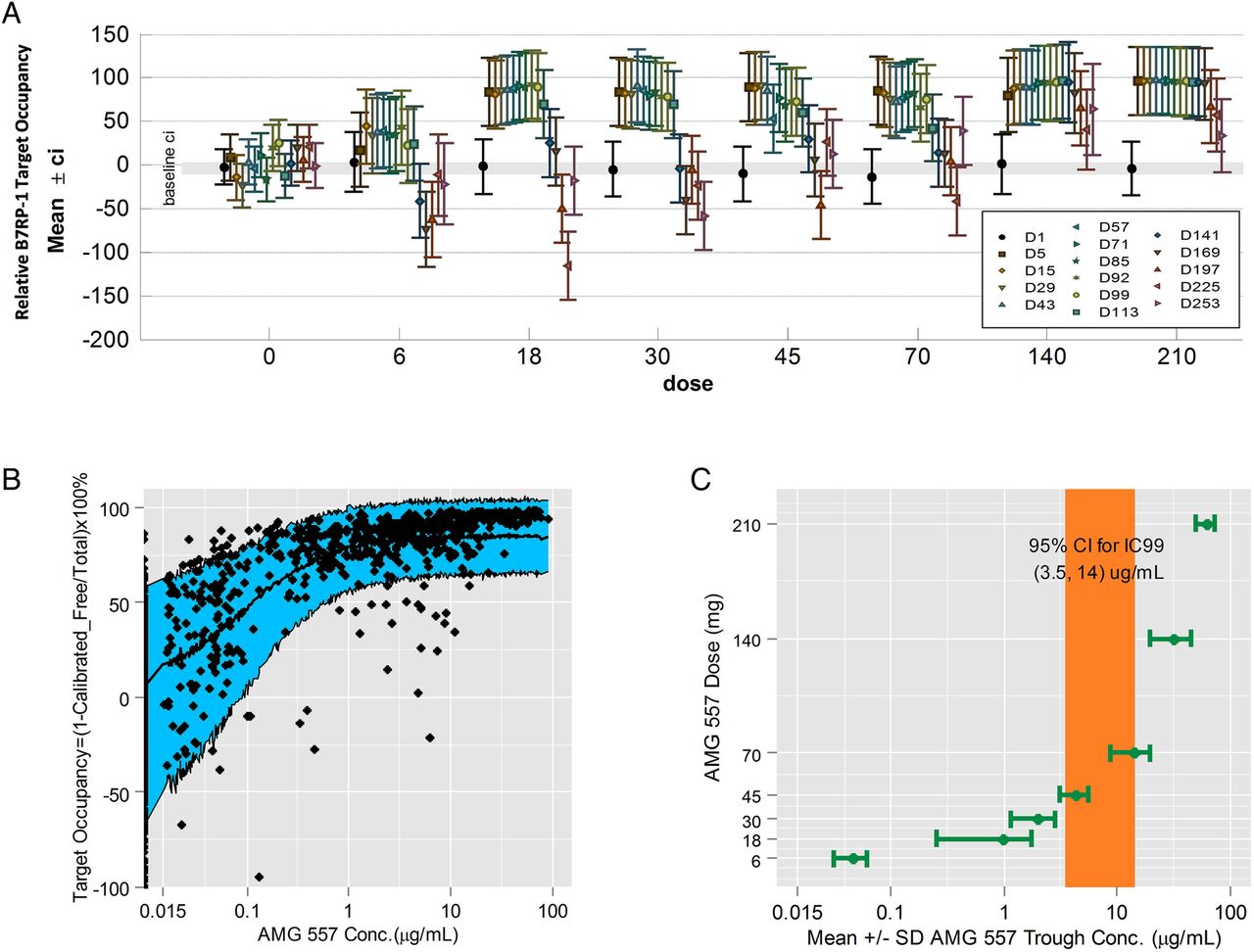
ICOSL target occupancy and the pharmacokinetics/pharmacodynamic relationship. (A) ICOSL target occupancy on circulating B cells from the multiple-ascending dose (MAD) study is shown by dose and visit (placebo: 0 mg and AMG 557: 6, 18, 30, 45, 70, 140 and 210 mg). Means from each applicable visit from day 1 to day 253 are graphed and coloured in chronological order within each dose group along with and 95% CIs. The baseline 95% CI of all data points is shown as a grey band. (B) The relationship between AMG 557 serum concentration and ICOSL target occupancy on circulating B cells from the combined single-ascending dose and MAD studies. ICOSL target occupancy is shown on the y-axis, and AMG 557 serum concentration on the x-axis. The observed data are shown as solid black symbols, and the model-predicted median and 80% prediction interval are shown as a solid line and blue shaded area. (C) Relationship between AMG 557 trough concentration at steady state with AMG 557 dose. The model-estimated IC99 (with 95% CI) for the AMG 557 target occupancy is shown as a coloured bar.
A simple inhibitory Emax model was used to correlate the ratio of free ICOSL over total ICOSL with AMG 557 serum concentration (figure 3B). The 95% CI for the IC99 was estimated to be between 3.5 and 14 μg/mL, which corresponded with near maximal occupancy of the target by AMG 557. The IC99 also corresponded to the apparent transition point between non-linear and linear pharmacokinetics (PK). Below the transition point, the drug exposure increased more than dose-proportionally when target-mediated disposition predominated. Above the transition point, which was achieved with 70 mg dose and above during active treatment at the steady state in the MAD study (figure 3C), the AMG 557 exposure increased approximately dose-proportionally consistent with a switch in clearance to the non-specific catabolism of IgG antibodies.43 ,44
ICOSL blockade reduces the anti-KLH IgG response
The effect of AMG 557 was assessed by measuring the antibody response to KLH. In the SAD study, subjects enrolled in the higher dose cohorts (60–210 mg subcutaneous dose groups) were immunised with two intradermal administrations of KLH, 4 weeks apart. The timing of the KLH administration varied by cohort. A protocol amendment changed the day of first injection of KLH by 1 week to allow for a higher concentration of AMG 557 at the time of immunisation. In the MAD study, subjects were immunised with KLH on days 57 and 85 (figure 1). Although all 56 subjects were planned per protocol in the MAD study to be immunised with KLH, 51 subjects were immunised and 5 were not immunised with KLH. Subjects that were not immunised with KLH were excluded from the anti-KLH IgG and anti-KLH IgM analyses. Missing data points from immunised subjects were not imputed, and means were based on available data.
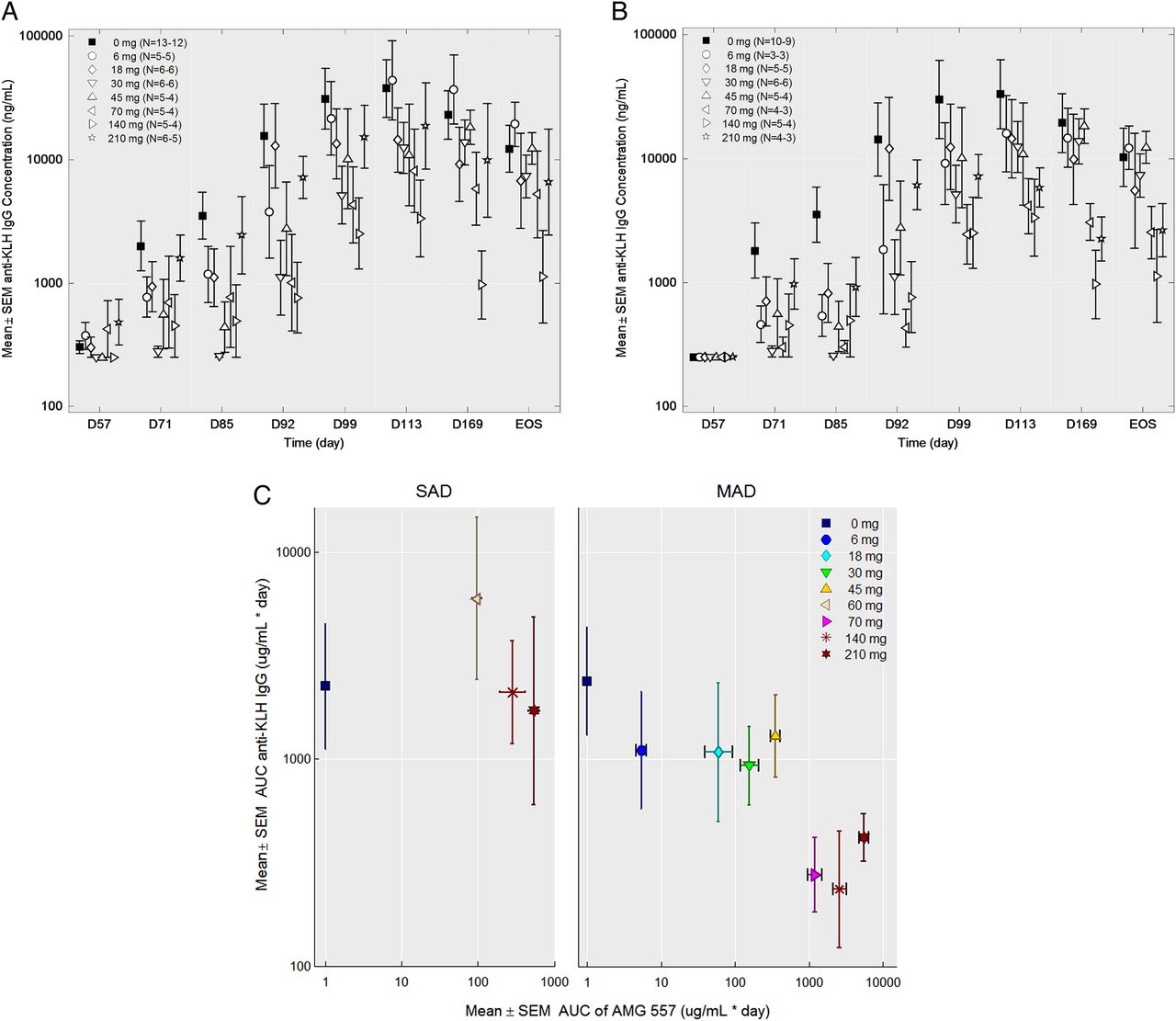
No statistically significant changes were seen in the anti-KLH IgM responses in either study (figure 4A,C). No statistically significant changes to anti-KLH IgG antibodies were observed in the SAD study (figure 4B). In the MAD study, the anti-KLH IgG antibody responses throughout the study period were significantly lower in the combined AMG 557-treated subjects compared with the combined placebo subjects from all the cohorts (p=0.0044) (figure 4D). Individual dose cohorts of AMG 557, compared with placebo, did not reach statistical significance. Also, 9 of the 56 subjects were found to have detectable levels (>250 ng/mL) of anti-KLH IgG antibodies prior to the first immunisation in the study, potentially confounding the assessment of primary and secondary responses to KLH in the study. A post hoc analysis was conducted excluding these subjects (figure 5), showing a clearer visual trend of a dose response. The reduction in KLH-specific IgG and lack of impact on the KLH-specific IgM was consistent with human and mouse genetic studies and in studies of pharmacological blockade in mice.11 ,13 ,14 ,42 An analysis of the relationship between the AUC) for AMG 557 serum concentrations and AUC for the anti-KLH IgG response is shown in figure 5C. This approach takes into account the impact of magnitude and duration of ICOSL blockade. Collectively, these analyses show a general dose response with maximal inhibition of the anti-KLH IgG response occurring at around 70 mg.
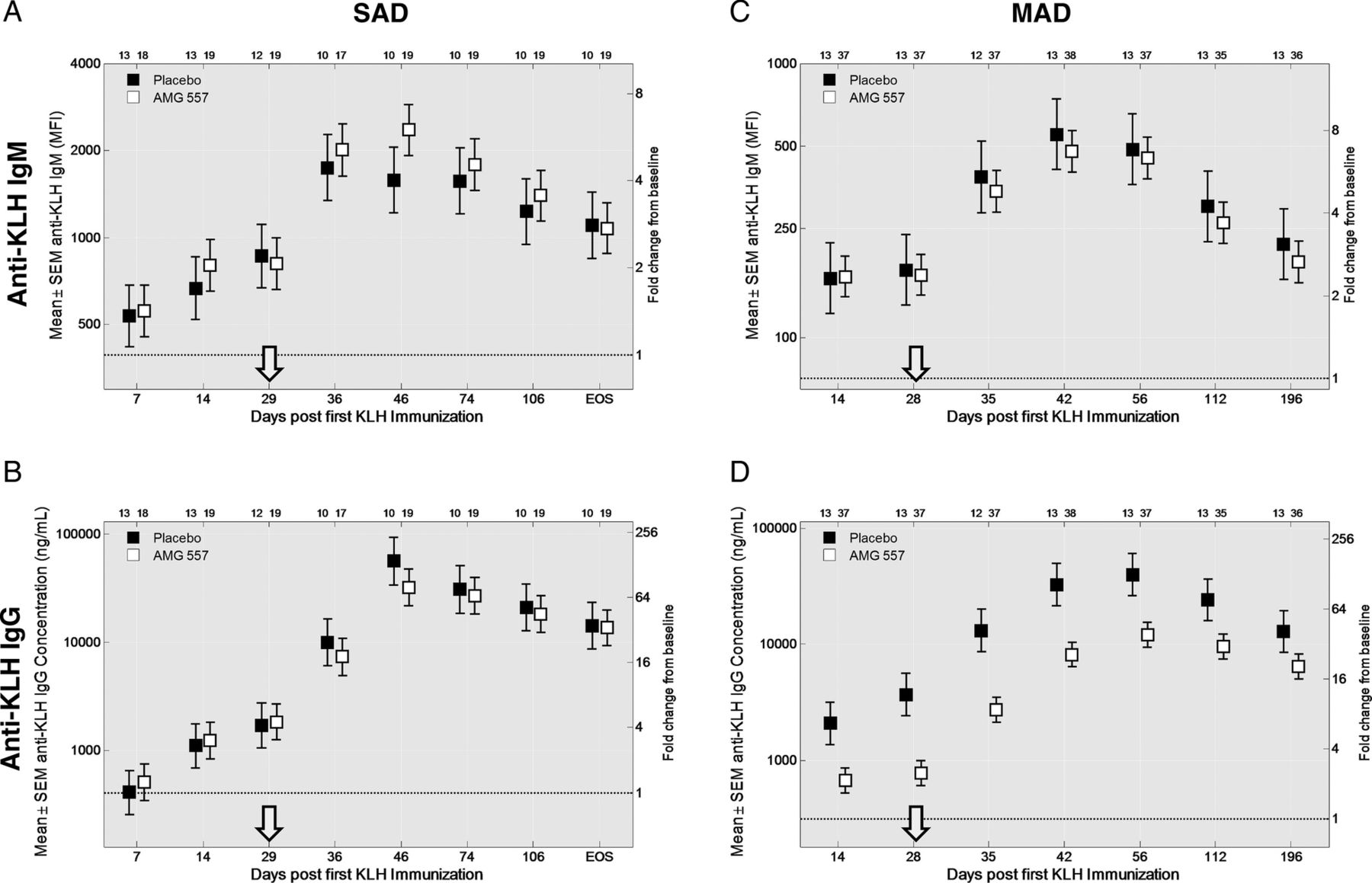
Pharmacodynamic effect of AMG 557—inhibition of the anti-keyhole limpet haemocyanin (KLH) IgG response. Anti-IgM and anti-IgG responses are shown for the placebo subjects and the aggregate AMG 557-treated subjects from the single-ascending dose (SAD) (A and B) and multiple-ascending dose (MAD) (C and D) studies. Data are shown for all four graphs by time following the first KLH immunisation on the x-axis and baseline-adjusted anti-KLH IgM (A and C) and anti-KLH IgG (B and D) values. The second KLH immunisation was administered approximately four weeks later (arrow). The fold change over baseline is shown on the right y-axis and the baseline-adjusted value (median fluorescence intensity (MFI) for IgM and concentration for IgG) is shown on the left y-axis; the dashed line indicates a fold change of 1. All AMG 557 doses were pooled for comparison to the placebo group. The number of values per symbol is indicated at the top of each graph. The plots show the mean±SEM.

Effect of dose level on the anti-keyhole limpet haemocyanin (KLH) IgG response in the multiple-ascending dose (MAD) study. (A) anti-KLH IgG serum values by individual cohort in the MAD study (uncensored). (B) Anti-KLH IgG serum values by individual cohort in the MAD study with the nine (of 51) pre-existing (ie, pre-immunisation) anti-KLH IgG-positive subjects removed. For both graphs, data are shown by study day (x-axis) and the anti-KLH IgG mean±SEM (y-axis), the dose level is indicated in the legend and the number of samples per symbol are shown in parentheses. (C) Mean area under the curve (AUC) (±SEM) of the serum anti-KLH IgG from the first KLH to 112 days after the first KLH (y-axis) is shown versus the mean AUC (±SEM) AMG 557 serum concentration from study day 1 to 112 days after the first KLH for the single-ascending dose (SAD) and the MAD studies. Pre-immunisation anti-KLH IgG-positive subjects from both studies were excluded from the analysis.
There were no apparent changes in tetanus antitoxin or total IgG levels observed throughout the study period (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serum anti-tetanus toxin IgG and total IgG levels do not change over time in the multiple-ascending dose study. Serum samples were tested at baseline (day 1, pre-dose), and post-dose days 85, 169 and 253 (end of study). Subjects are grouped by dose level. (A) Anti-TT IgG levels. For samples with tetanus antitoxoid concentration >8.30 IU/mL (the upper limit of the assay), 8.30 IU/mL was used for the analysis. (B) Total IgG levels. The number of samples tested is indicated in parentheses in the figure legend.
Impact of AMG 557 administration on circulating lymphocyte subpopulations and SLE-associated measures
A comprehensive panel of cell surface markers was implemented to measure the potential impact of ICOS pathway blockade on lymphocyte subpopulations. These populations included T, B and NK cells, and select subpopulations including memory Tfh cells (CD45+CD3+CD4+CD45RO+CXCR5+), Treg cells (CD45+CD3+CD4+CD25+CD127−), B cell subsets (CD45+CD19+CD20+: cB1 IgD+CD27−, cB2 IgD+CD27+, cB3 IgD+CD27−) and plasmablasts/plasma cells (CD45+CD19+: CD27+CD38++). No clear changes were observed in any of these defined subpopulations as a result of AMG 557 administration (data not shown).
No discernible changes were observed in lupus serologies, complement levels or disease measures (BILAG, SELENA-SLEDAI) (data not shown).
Discussion
The ICOS:ICOSL pathway represents a promising target for therapeutic immunomodulation of autoimmune diseases. Immunological investigations in mice and humans, including analyses of ICOS null individuals, have detailed the role of this co-stimulatory pathway in driving antigen-specific T cell responses. The importance of ICOS for the development of T cell subsets such as Tfh and Th17 cells and for driving class-switched antibody responses implicates it as a potential key mediator of autoimmune diseases. This is the first report of pharmacological blockade of the ICOS:ICOSL pathway in humans.
Consistent with other therapeutics targeting a cell surface molecule, the AMG 557 PK profile showed non-linear characteristics at lower dose levels. AMG 557 concentration increased greater than dose-proportionally over the dose range of 1.8–140 mg subcutaneous in the SAD study and 6–70 mg subcutaneous in the MAD study. Flow cytometric analyses of ICOSL target occupancy on circulating B cells indicated an IC99 between 3.5 and 14 μg/mL using a simple Emax model. Within the IC99 serum concentration range, a transition from target-mediated (ie, non-linear) elimination to linear elimination occurred, likely suggesting ICOSL saturation in blood as well as compartments not measured (or accessible).
In the MAD study, AMG 557 significantly reduced the anti-KLH IgG response, but a dose–response to AMG 557 was not readily discernible given the high degree of variability in the data. Also, 9 of the 51 subjects immunised with KLH had detectable levels of anti-KLH IgG (eg, >250 ng/mL) prior to KLH immunisation. Parallel validation of the anti-KLH IgG assay from independent samples revealed that 16% of healthy volunteers and 12% of subjects with SLE had detectable levels (eg, >250 ng/mL) of serum anti-KLH IgG. Preincubation with increasing concentrations of KLH reduced the anti-KLH IgG signal, indicating KLH specificity in the absence of intentional KLH immunisation, and possibly cross-reactivity. Protective cross-reactivity between shared carbohydrate epitopes on KLH and Schistosoma mansoni has been described in rats.45 Regardless of the origin of the KLH-binding IgGs in the baseline samples of this study, the KLH may in fact represent a ‘booster antigen’ and not a ‘neoantigen’, as intended. A post hoc analysis excluding the preimmunisation-positive subjects in the MAD anti-KLH IgG data set appeared to clarify the dose–response relationship in the overall dataset. Further evaluation of the AUC of anti-KLH IgG levels as a function of the AUC of AMG 557 serum concentration clearly shows a visual trend towards a dose–response with multiple doses of 70 mg subcutaneous as the inflection point of the curve. This inflection point correlates well with the steady-state trough concentrations of AMG 557 that were above the IC99 concentration for target occupancy.
Beyond KLH, ICOSL blockade in subjects with SLE may reduce subclinical inflammation and GC activity more broadly, including those that harbour autoreactive B cells. Bystander cell expression of ICOSL has also recently been shown in mice to be required for ICOS-mediated trafficking to form GCs.46 These data will provide helpful information to guide dose selection for subsequent efficacy studies.
As expected for subjects with SLE with mild, stable disease, there were no discernible changes in disease activity (BILAG, SELENA-SLEDAI), lupus serology, complement levels or circulating lymphocyte subpopulations (data not shown). While the percentage of subjects with a positive ANA at baseline in the MAD study was surprisingly low, a documented history of a positive ANA (titre >1:80) was required for enrolment. Additionally, SLE-associated serum biomarkers (including IP-10) as well as ICOS levels on T cells were elevated at baseline (data not shown), consistent with other reports in SLE.
The safety profile of AMG 557 appeared acceptable in these early and small studies, but more data are needed, particularly with longer-term administration. There was no evidence of cellular depletion or of aberrant activation as a result of AMG 557 administration. ICOSL is detectable predominantly on the surface of APCs, but it can be upregulated on endothelial cells and some epithelial cells, and mRNA can be induced on testes, kidney and peritoneum.47 ,48 Additionally, ICOSL has been found on human placental trophoblast cells.49 We also note that the 15 described ICOS null patients11–14 ,50 ,51 are characterised by low-circulating memory B cells, low-memory TFH, low IgG, low IgA and manifest clinically with opportunistic infections and colitis.
Recent advances in targeted therapies for SLE have validated the cytokine BAFF as a key driver of disease.52 Numerous other targets are currently being pursued in clinical trials for SLE and related autoimmune conditions.31 The overall safety, PK and pharmacodynamic biomarker results of these reported phase I studies in patients with SLE establish ICOSL as a viable target for pharmacological intervention and support further evaluation of AMG 557 as a therapeutic for SLE and other autoimmune diseases.
Acknowledgments
The authors thank Drs Brian Kotzin, Steven Zoog and Chris Russell for input and helpful scientific discussions. They also to thank Dr Joan Merrill for her role in the SLEDAI and BILAG training, safety reviews and overall advice on these studies. Finally, they also wish to thank the investigators and patients for study participation.
References
Footnotes
Contributors BAS carried out the flow cytometry, receptor occupancy, cytometric bead array and anti-TT studies and drafted the manuscript. WT was the medical monitor for the MAD study and helped to revise the manuscript. AK participated as an investigator and helped design both studies and helped to revise the manuscript. JP analysed the serum AMG 557 concentrations and anti-KLH IgG and IgM analyses and helped to revise the manuscript. GEA analysed and graphed the anti-KLH IgG and IgM and anti-TT IgG data and helped revise the manuscript. MJB participated in the statistical analysis of the flow cytometry, KLH and TT analyses and helped revise the manuscript. KC was the study manager for both studies and helped revise the manuscript. CLG carried out the flow cytometry and receptor occupancy analysis and helped revise the manuscript. AK carried out the anti-AMG 557 antibody testing and helped to revise the manuscript. CW performed statistical analyses on patient demographics and standard biomarkers (anti-dsDNA) and helped to revise the manuscript. JF carried out cytometric bead array assays to measure anti-KLH IgG and IgM, participated in the study design and helped to revise the manuscript. JBC designed the two studies, oversaw the study execution, oversaw the PK, anti-AMG 557 antibody and biomarker sample analysis and helped revise the manuscript. All authors read and approved the final manuscript.
Funding This work and the study presented herein were financially funded by Amgen, Thousand Oaks, California, USA, and MedImmune, Gaithersburg, Maryland, USA. AMG 557 is being co-developed by MedImmune (a wholly owned subsidiary of AstraZeneca) and Amgen and may be referred to as AMG 557 or MEDI5872.
Competing interests All authors are either employees and shareholders or contractors of Amgen or consultants who have received research support from Amgen.
Ethics approval See online supplementary appendix A.
Provenance and peer review Not commissioned; externally peer reviewed.