Article Text

Abstract
Objectives To compare the pharmacokinetics (PK), safety and tolerability of subcutaneous (SC) and intravenous anifrolumab, an anti–type I interferon receptor monoclonal antibody in development for SLE, in healthy volunteers.
Methods In this Phase I randomised, placebo-controlled study, 30 adults were assigned to three treatment cohorts (anifrolumab 300 mg SC (n=6), anifrolumab 300 mg intravenous (n=6), anifrolumab 600 mg SC (n=6)) and placebo (n=4/cohort). Serial blood samples were collected up to Day 84 to measure anifrolumab concentrations and antidrug antibodies (ADAs). PK parameters were estimated by noncompartmental analysis.
Results Maximum serum concentrations in SC cohorts occurred after 4–7 days. Anifrolumab serum concentrations were below the limit of detection in all individuals by Day 84. Exposure to SC anifrolumab increased dose proportionally from 300 mg to 600 mg based on area under the serum concentration-time curve. Anifrolumab 300 mg SC exposure reached 87% of the intravenous exposure. Anifrolumab 300 mg SC and placebo administration elicited minimal injection-site reactions. Transient injection-site induration occurred in five of six individuals after anifrolumab 600 mg SC and two of four individuals after placebo. Transient, mild to moderate injection-site induration and pruritus occurred simultaneously in two of six individuals after anifrolumab 600 mg SC. Adverse events were reported by 50% (n=9) of anifrolumab-treated individuals and 33% (n=4) of placebo-treated individuals. ADAs were detected in only one individual in the anifrolumab 300-mg intravenous group at the Day 84 assessment.
Conclusion Anifrolumab 300-mg SC exposure was 87% of intravenous administration, with single SC anifrolumab administrations well tolerated in healthy volunteers.
- systemic lupus erythematosus
- biologics
- pharmacokinetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
SLE is a chronic, multisystem autoimmune disease with extreme heterogeneity between patients with respect to clinical manifestations, organ involvement and disease severity.1 The incidence and prevalence of SLE are estimated to be as great as 32 cases per 100 000 individuals per year and 520 cases per 100 000 individuals, respectively.1 Although the 5-year survival rate has increased to almost 90% in the last 70 years, there remains an unmet need for therapeutics that achieve stable low disease activity, prevent ongoing organ damage and reduce the need for high-dosage corticosteroid use.2
Increased clarity of the pathogenesis of SLE is driving development of immunotherapy targeted to specific patient endotypes. A hallmark of SLE is the presence of self-reactive autoantibodies bound to circulating nuclear antigens, which drive cellular overproduction of type I interferon (IFN) and other proinflammatory cytokines.3 A substantial body of evidence supports an important role of type I IFNs in the pathogenesis of SLE.4–9 The successful development of anti–type I IFN therapeutics is anticipated to advance the medical management of patients with SLE significantly.
Anifrolumab, a fully human immunoglobulin G1 (IgG1) monoclonal antibody targeting type I IFN alpha receptor 1 (IFNAR1), is in Phase III development as an intravenous therapeutic for moderate to severe SLE. Anifrolumab inhibits type I IFN-dependent cell signalling by binding to IFNAR1 and blocking formation of the IFN/IFNAR complex.10 In the Phase IIb MUSE trial (NCT01438489), anifrolumab 300 mg every 4 weeks intravenously plus standard of care significantly decreased SLE disease activity across a range of endpoints, especially for patients with type I IFN gene signature test high results at baseline.7 Two Phase III trials (TULIP-I/II; NCT02446899/NCT02446912) are currently ongoing to further evaluate the efficacy and safety of intravenous anifrolumab for patients with moderate to severe SLE.
Of the three novel anti–type I IFN antibodies (rontalizumab, sifalimumab and anifrolumab) evaluated in Phase II clinical trials for SLE, the intravenous formulation of anifrolumab had the highest efficacy relative to placebo.7 11 12 However, patients and health care providers generally prefer subcutaneous (SC) over intravenous administration of biologics.13–18 Thus, we profiled the pharmacokinetics (PK), safety and tolerability of anifrolumab administered SC and intravenously to healthy volunteers.
Patients and methods
Population, study design and objectives
This was a Phase I, single-centre (Baltimore, Maryland, USA), double-blind, randomised, placebo-controlled trial (NCT02601625) involving healthy volunteers. The trial included individuals aged 18–55 years with a body mass index of 18–32 kg/m2 and body weight ≥50 kg. Additional inclusion criteria comprised a normal Pap smear for female volunteers, sufficient abdominal adipose tissue for SC injection, no prior or current latent or active tuberculosis and no history of recent or severe Herpes zoster infection. Exclusion criteria included any recent infection requiring hospitalisation or treatment with parenteral antimicrobials, any severe herpes infection at any time prior to dosing and receipt of a recent live or attenuated vaccine administration.
The study consisted of an enrolment and screening period of up to 28 days, a 3-day residential period and follow-up visits on Days 4, 7, 10, 14, 21, 28, 42, 56 and 84 (figure 1). Volunteers were assigned to three sequential treatment cohorts of equal size and randomised within each cohort to receive a single dose of either anifrolumab (n=6/cohort) or placebo (0.9% normal saline; n=4/cohort). For Cohort 1 (n=10), anifrolumab 300 mg or placebo was administered as two separate 1-mL SC injections via syringe and 27-gauge 1/2" needle. For Cohort 2 (n=10), anifrolumab 300 mg or placebo was administered as a 100-mL intravenous infusion over 30 min. For Cohort 3 (n=10), anifrolumab 600 mg or placebo was administered as 4 mL SC by large-volume infusion pump (Perfusor Space Infusion Pump, B. Braun Medical, Inc.). SC injectates were administered in the anterior abdominal wall while avoiding a 5-cm radius around the umbilicus.
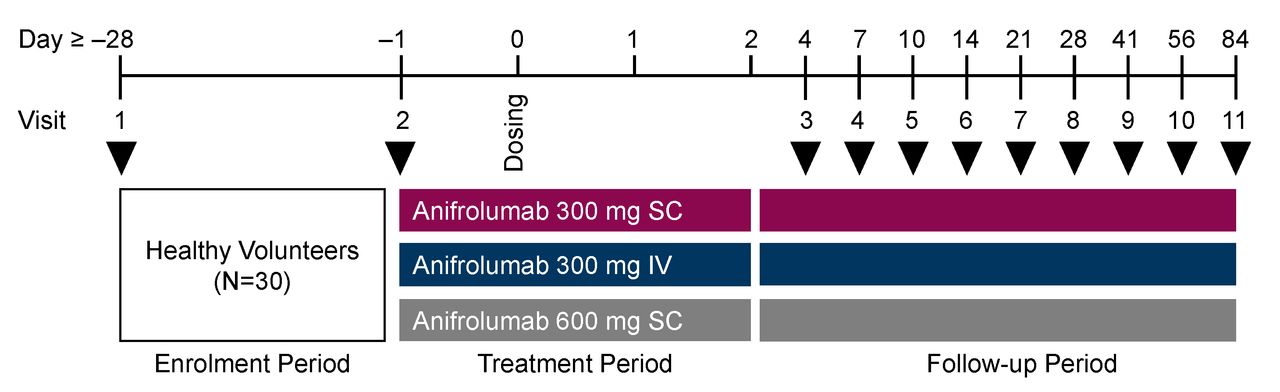
Study design. IV, intravenous; SC, subcutaneous. Black arrow heads refer to visits.
Pharmacokinetics, safety, tolerability and immunogenicity
The objective of this study was to evaluate the PK, safety, tolerability and immunogenicity of single SC and intravenous administrations of anifrolumab. Serial venous blood samples were collected pre dose, 5 min after SC injection or intravenous infusion ended, 24 and 48 hours post dose and at Visits 3–11. Samples were analysed to measure serum anifrolumab concentrations with a validated assay, as previously described.19 Serum from blood samples collected pre dose and at Visits 3, 8 and 11 was evaluated also for the presence of anti-drug antibodies using a validated bioanalytical method.19
Safety endpoints included adverse events, adverse events of special interest, laboratory assessments and vital signs. Adverse events of special interest were serious infections, opportunistic infections, anaphylaxis, malignancy, Herpes zoster, tuberculosis (including latent tuberculosis), influenza, vasculitis and major adverse cardiac events (including stroke, myocardial infarction, or cardiovascular death).
Tolerability endpoints included assessments of the SC injection sites for pain (100-mm visual analogue scale (VAS)), pruritus (100-mm VAS), erythema (largest diameter, mm) and induration (largest diameter, mm). Injection-site assessments occurred at 10, 20 and 30 min and then 1, 2, 4, 8, 24 and 48 hours post injection. Injection-site reactions were not recorded as adverse events unless an individual complained of these at any time point post injection.
Statistical analysis
The safety analysis set included all individuals who received at least one dose of anifrolumab or placebo. The PK analysis set included all individuals in the safety analysis set for whom at least one primary PK parameter could be calculated. Data were summarised by descriptive statistics. PK parameters were calculated by non-compartmental analysis with Phoenix WinNonlin V/6.2 (Certara, Inc., Princeton, New Jersey, USA) and included the area under the serum concentration-time curve (AUC), clearance (CL, CL/F), maximum serum concentration (Cmax) and time to reach maximum serum concentration (tmax). All data were analysed with SAS System V.9.2 (SAS Institute, Inc., Cary, NC, USA) .
Results
Volunteer disposition
Thirty adult healthy volunteers were randomised to and completed treatment, and 28 (93%) individuals completed the study (table 1). Individuals had a median age of 30 years (range: 19–55 years), and a mean body mass index of 25.4 kg/m2 (SD: 3.0 kg/m2). Most individuals were black or African-American (63%), and more than half of the volunteers were male (63%). Two individuals were lost to follow up: one who received placebo in the anifrolumab 600-mg group after Visit 6 (Day 14) and one who received anifrolumab in the anifrolumab 600-mg group after Visit 7 (Day 21). All 30 individuals were included in the PK and safety analyses sets.
Demographics and baseline characteristics
Anifrolumab pharmacokinetics
In the SC cohorts, Cmax (mean (SD)) was 36.2 (11.6) μg/mL in the anifrolumab 300-mg group and 63.9 (20.5) μg/mL in the anifrolumab 600-mg group. Peak serum concentrations (Tmax) occurred 4–7 days after injection (table 2 and figure 2). Exposure to SC anifrolumab increased approximately dose proportionally from 300 mg to 600 mg based on mean (SD) AUC (anifrolumab 300 mg: 785 (331) day·μg/mL; anifrolumab 600 mg: 1828 (680) day·μg/mL; table 2). At the 300-mg dose, anifrolumab exposure after SC administration reached approximately 87% of the intravenous administration exposure (mean AUC (SD): 907 (175) day·μg/mL). All individuals had quantifiable serum anifrolumab concentrations in all samples from the time of dosing until at least 28 days post dose. Anifrolumab serum concentrations were below the limit of detection for all individuals by 84 days post dose (figure 2).
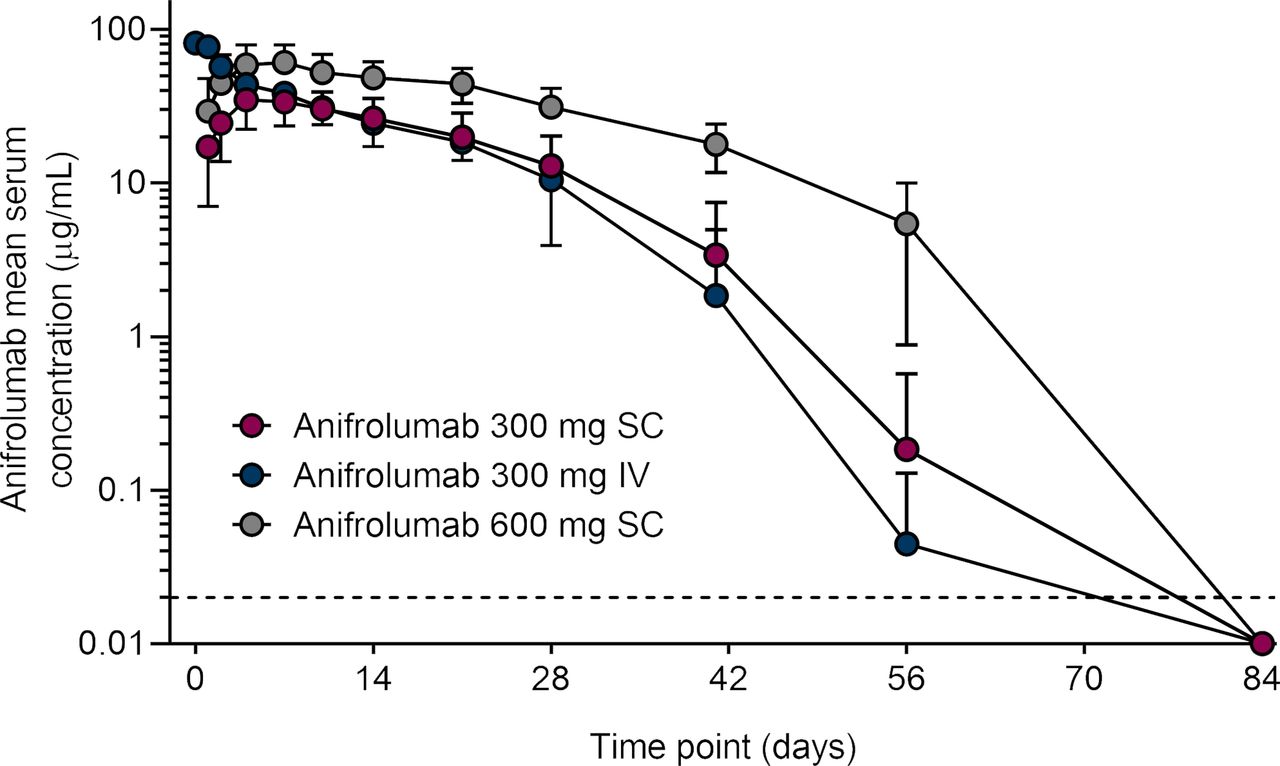
Mean serum concentration time profiles of anifrolumab following subcutaneous and intravenous administration of anifrolumab.a aData below the limits of detection are plotted as one-half of the lower limits of quantification (0.02 µg/mL; dashed line). Values are means and SD. IV, intravenous; SC, subcutaneous.
Anifrolumab serum PK parameters following subcutaneous and intravenous administration of anifrolumab
Safety, tolerability and immunogenicity
Adverse events were reported by 50% (n=9) and 33% (n=4) of anifrolumab-treated and placebo-treated individuals, respectively (table 3). The most common adverse events in anifrolumab-treated individuals were upper respiratory tract infection (n=3; 17%) and dry throat (n=2; 11%). No serious adverse events were reported (table 3). Anti-drug antibodies were detected in only one individual in the anifrolumab 300-mg intravenous group at the Day 84 assessment.
Number of volunteers who had at least one adverse event
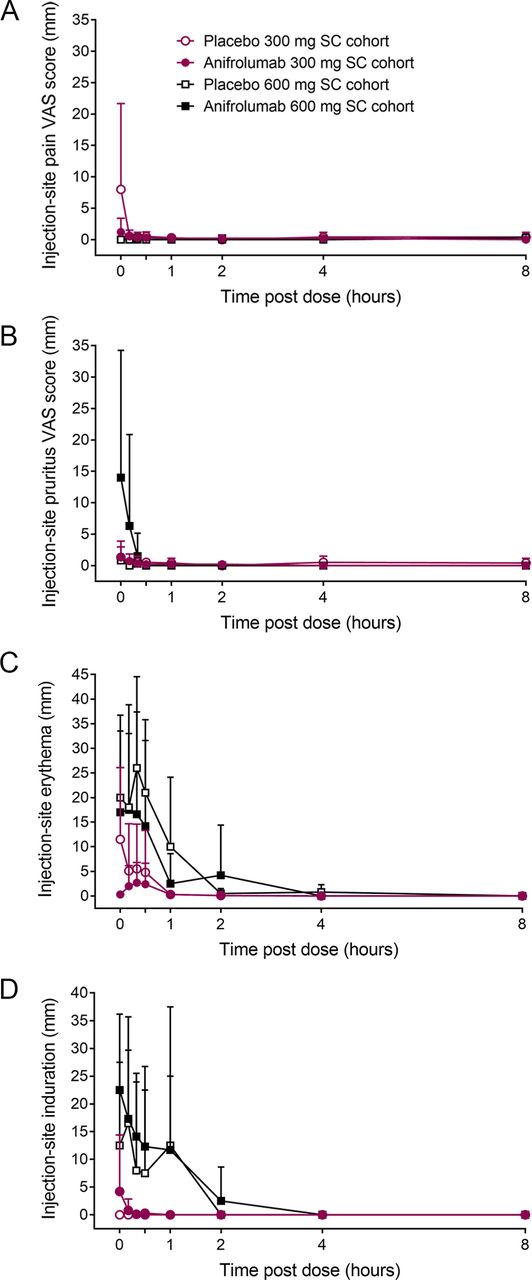
Injection-site pain was either absent (600-mg cohort) or minimal to mild (300-mg cohort) and resolved within 10 min after administration (figure 3A). Injection-site pruritus was minimal after administering anifrolumab or placebo in the 300-mg SC cohort and was mild to moderate in two individuals who received anifrolumab 600 mg (figure 3B). Pruritus resolved by 20 min post dose in both cohorts. Injection-site erythema immediately after SC dosing was more pronounced in individuals receiving the larger volume infusion (600-mg cohort) compared with those receiving the SC injection (300-mg cohort; figure 3C). There were minimal differences in erythema size between anifrolumab and placebo groups in the 600-mg cohort. Erythema resolved within 8 hours post dose in all cohorts. Injection-site induration was more pronounced immediately after 600-mg SC infusions than after 300-mg SC injections (figure 3D). Induration resolved in all groups by 4 hours post dose. Induration was not associated with pain or erythema.
{kind=link}
{kind=link}
{kind=link}
Injection-site reactions after subcutaneous administration of anifrolumab and placebo. Values are means and SD. SC, subcutaneous; VAS, visual analogue scale.
Discussion
Unresolved medical needs, such as insufficient disease control and toxicity of current therapies, and a better understanding of the pathobiology of lupus have stimulated development of endo-type-specific therapeutics over the past decade. More than 25 novel agents are undergoing Phase II/III clinical trials for the treatment of SLE (https://clinicaltrials.gov/). Antibodies targeting the type I IFN pathway, rontalizumab, sifalimumab and anifrolumab, have recently been assessed in Phase II randomised controlled trials.7 11 12 Primary endpoints were not met in the rontalizumab trial but were reached in the sifalimumab trial. More patients (58.3%) receiving 200-mg sifalimumab intravenously achieved an SLE Responder Index of 4 points (SRI(4)) at Day 365 compared with those receiving placebo (45.4%). The anifrolumab trial also reached its primary endpoint of an SRI(4) and sustained oral corticosteroid reduction at Day 169. This composite endpoint was achieved by 34.3% of patients in the 300-mg anifrolumab intravenous group compared with 17.6% of patients receiving placebo. A similar difference in SRI(4) was observed at Day 365 (300-mg anifrolumab intravenous: 51.5% vs placebo: 25.5%). Although these antibodies have not been compared side by side in a single trial, the data suggest that intravenous anifrolumab has best-in-disease efficacy for this class of drug in patients with moderate to severe SLE.20 The intravenous formulation of anifrolumab is undergoing further evaluation in Phase III trials (NCT02446899 and NCT02446912).
In the past 50 years, only one new therapeutic, belimumab, has been approved for the treatment of SLE. The approval of intravenous anifrolumab would be a significant advancement in the management of SLE, and the potential for SC administration would be further advantageous because patients and health care providers prefer SC over intravenous injections.17 Several studies have evaluated patients’ preference after receiving biologicals via both intravenous and SC routes of administration.13 14 16 18 21 Patients cited time saving, convenience and avoidance of venipuncture as the primary reasons they preferred the SC route of administration. The presence of transient, mild to moderate injection-site reactions after SC, but not intravenous, administration of a biologic was not a sufficient deterrent.
Notwithstanding the evidence that patient preference and pharmacoeconomics are the primary drivers of SC formulation development, initial studies are required to demonstrate that the SC route of administration has comparable drug exposure, safety, efficacy and tolerability to an intravenous formulation.22 Accordingly, we evaluated the PK, safety and tolerability of SC and intravenous administration of anifrolumab for healthy volunteers. We found that the exposure of anifrolumab 300 mg SC was approximately 87% of intravenous administration and that SC anifrolumab was well-tolerated and with minimal injection-site reactions. In our study, the incidence and magnitude of these reactions were consistent with prior studies.13 14 16 18 21
In general, the pharmacokinetic properties of monoclonal antibodies are markedly different to most small molecule drugs.23 The larger molecular size of antibodies contributes to slower absorption from SC injection sites via the lymphatics, slower distribution to tissues and lower volumes of distribution. If there is significant presystemic catabolism of the subcutaneously administered antibody, bioavailability is often low to intermediate.24 The times to peak serum concentration and bioavailabilities of registered IgG1 monoclonal antibodies marketed as SC formulations typically range from 3 to 8 days and 60%–80%, respectively.25 The tmax for anifrolumab after SC administration is consistent with this range. In contrast, anifrolumab exposure after SC administration suggests that the bioavailability of the antibody may be relatively high. Collectively, our data support the further development of the SC formulation of anifrolumab. The pharmacokinetics, pharmacodynamics and safety of multiple SC administrations of anifrolumab are currently being evaluated in a Phase II trial (NCT02962960). This placebo-controlled study is assessing two fixed dosages of anifrolumab (150 mg and 300 mg) administered subcutaneously every 2 weeks for up to 50 weeks, within a treatment period of 52 weeks. The objective of the study is to identify the most appropriate SC dosage for a future Phase III trial.
A limitation of our study was the absence of pharmacodynamic assessments after SC and intravenous administration of anifrolumab to healthy volunteers. The primary pharmacodynamic effect of anifrolumab in patients with SLE is suppression of the type I IFN gene signature. Increased type I IFN expression initiates multiple signal transduction pathways, which in turn lead to expression of up to 2000 IFN-stimulated genes.26 Anifrolumab 300 mg intravenously administered to patients with moderate to severe SLE every 4 weeks for 48 weeks suppressed the type I IFN gene signature with a median neutralisation range of 85%–90% from Day 29 to Day 365.27 The IFN gene signature is relatively low in healthy volunteers, precluding assessments of anifrolumab pharmacodynamics in our study.28 Additional limitations were that the volunteers were mostly black or African-American and male. The pharmacodynamics of SC anifrolumab will be assessed in patients with SLE during the Phase II clinical trial described earlier.
In conclusion, SC administration of anifrolumab 300 mg and anifrolumab 600 mg exhibited dose-proportional pharmacokinetics in healthy volunteers. Anifrolumab exposure after SC administration was relatively high compared with similar studies of registered IgG1 monoclonal antibodies marketed as SC formulations. Anifrolumab, administered as an intravenous infusion or an SC injection, was well tolerated in healthy subjects. Collectively, these data support the further development of anifrolumab as an SC formulation for the treatment of patients with lupus disorders.
Acknowledgments
Some of these data have been published in abstract form following presentation at the 2017 Annual Meeting of the American College of Rheumatology (Tummala R, Rouse T, Berglind A, Santiago L. Arthritis Rheumatol. 2017; 69 (suppl 10)). Editorial support was provided by Francis J Golder, BVSc, PhD, of JK Associates, Inc., and Michael A Nissen, ELS, of AstraZeneca. This support was funded by AstraZeneca.
References
Footnotes
Contributors RT proposed the study design. LS performed the pharmacokinetics analysis. TR and AB performed the statistical analyses. All authors contributed to developing the manuscript.
Funding This study was funded by AstraZeneca.
Competing interests RT, TR and AB are employees of AstraZeneca. LS is an employee of MedImmune LLC and a shareholder of AstraZeneca.
Patient consent All volunteers provided informed consent before the start of the study.
Ethics approval The clinical protocol was approved by the institutional review board and independent ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The study protocol is available upon request. Study data are available upon request and completion of a data sharing agreement with AstraZeneca via the Data Request Portal (https://astrazenecagroup-dt.pharmacm.com//DT/Home/Index/).