Article Text

Abstract
Objectives Activation of endosomal toll-like receptor (TLR)7 or TLR9 has been proposed as a critical step for the initiation and development of SLE. Traditional spontaneous lupus models normally introduce multiple risk alleles, thereby adding additional confounding factors. In the induced lupus models, the role of TLR9 remains unclear. In the present study, we explored the role of an imbalance between TLR7 and TLR9 pathways in the pathogenesis of lupus and its associated vasculopathy using the imiquimod model in TLR9 KO/B6 background.
Methods Wild type (WT) and Tlr9-/- mice were epicutaneously treated with imiquimod cream 5% on both ears three times per week for indicated times. At euthanasia, mice were analysed for organ involvement, endothelium-dependent vasorelaxation, serum autoantibodies, and innate and adaptive immune responses.
Results Compared with the lupus-like phenotype that develops in imiquimod-treated WT mice, Tlr9-/- mice exposed to imiquimod have increased severity of autoimmunity features and inflammatory phenotype that develops at earlier stages. These abnormalities are characterised by enhanced TLR7 expression and immune activation, increased immune complex deposition, Th1 T cells and dendritic cell kidney infiltration and significant impairments in endothelial function. Modulation of TLR7 expression was observed in the Tlr9-/- mice.
Conclusions These findings further underscore the protective role of TLR9 in TLR7-driven autoimmunity and also in the development of vasculopathy, further strengthening the importance of tightly manipulating TLRs in putative therapeutic strategies. This study provides a new model of accelerated lupus phenotype driven by danger-associated molecular patterns.
- systemic lupus erythematosus
- autoimmunity
- inflammation
- cardiovascular disease
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
SLE is an autoimmune disorder characterised by profound dysregulation of innate and adaptive immune responses with aberrant production of proinflammatory cytokines, generation of antinuclear antibodies and multiorgan damage.1 Subjects with SLE are also at a significant risk of cardiovascular complications from accelerated atherosclerosis largely driven by immune dysregulation.2 As nuclear autoantigens (including double-stranded DNA (dsDNA), Sm, RNP and Sm-RNP) are targeted in SLE, activation of endosomal toll-like receptors (TLR) 7 or 9 has been suggested as a critical step for lupus initiation and development, through the induction of type I interferons (IFN) and other proinflammatory cytokines. TLR7 and TLR9 signalling induced by single-stranded RNA-associated and unmethylated CpG DNA-associated autoantigens, respectively, can activate dendritic cells (DC) and self-reactive B cells to break self-tolerance.3 Various TLR7-deficient or TLR9-deficient lupus-prone mouse models have demonstrated that TLR7 is critical in generation of anti-Sm and anti-RNA autoantibodies, while TLR9 is indispensable for producing antichromatin and antinucleosome autoantibodies.4–7
Counterintuitively, despite the absence of antinucleosome and antichromatin autoantibodies, the absence of TLR9 in several spontaneous murine models of SLE can lead to a phenotype characterised by exacerbation of lupus manifestations and immune dysregulation. These models include the B6-lpr/lpr-Tlr9-/-, TLR9-deficient B6.Nba2 and TLR9-deficient lupus-prone FcγRIIB-deficient mice.7–9 A major drawback of the spontaneous lupus models is its polygenic (disease is driven by multiple alleles, such as MRL/faslpr model) or monoallelic nature (disease is driven by single alleles, such as B6.yaa). For example, the lpr mutation in Fas in the MRL/fas lpr model may interfere with T cell activation, proliferation and cytokine/chemokine production, thereby adding additional confounding factors to the effect of TLR9 alone.10
Of note, in the induced lupus models, the role of TLR9 remains unclear. In the pristane-induced lupus model on Balb/c background, deficiency of TLR9 accelerates lupus symptoms and autoantibody synthesis.11 However, when the pristane model was on B6 background, either no differences between Tlr9-/- mice and control mice were observed,12 or improvement in clinical disease and inflammation were evident.13
Recently, an inducible lupus model that uses epicutaneous application of TLR7 agonists has been described in several genetic backgrounds, including B6 mice.14 This model, which resembles the phenotype of the TLR7 transgenic (TLR7tg) lupus-prone mice,15 16 leads to several phenotypic and functional changes characteristic of human SLE, including ANA development, renal immune complex (IC) deposition, type I IFN responses and immune activation.14 In the present study, we explored the role of an imbalance between TLR7 and TLR9 pathways in the pathogenesis of lupus using the imiquimod model in Tlr9-/-/B6 background when compared with wild-type (WT) mice. We found that imiquimod-treated Tlr9-/- mice have accelerated autoimmunity and heightened inflammatory phenotype, suggesting a protective role of TLR9 in TLR7-driven autoimmunity.
Results
Imiquimod-induced autoimmunity is exacerbated in the absence of TLR9
We first compared the immune responses between untreated WT mice and untreated Tlr9-/- mice, and did not notice significantly differences in terms of spleen size, total splenocyte numbers, immune cell composition or cytokine productions in the spleen or kidney IC deposition (online supplementary figure 1). We initially exposed mice to imiquimod for up to 5 weeks, as previously reported.14 In the original description of this model, WT mice exposed to imiquimod began to die after 8 weeks of treatment.14 However, Tlr9-/- mice exposed to imiquimod for more than 3 weeks showed accelerated death rates and profound weight loss, indicating an accelerated phenotype (online supplementary figure 2). Indeed, the spontaneous death rate was approximately 20% in Tlr9-/- mice in 4-week imiquimod treatment and approximately 40% in 5-week imiquimod treatment, while approximately 8.3% WT mice died after 5-week imiquimod treatment (online supplementary figure 2A). Given the high incidence of animal death, we examined the phenotype at earlier time points, 3 weeks after initiation of imiquimod. The imiquimod-induced lupus model is characterised by development of splenomegaly in WT mice.14 While both groups showed enhanced spleen size at 3 weeks of treatment, imiquimod-treated Tlr9-/- mice displayed significantly increased splenomegaly and splenocyte numbers compared with imiquimod-treated WT mice, with no evidence of sexual dimorphism (figure 1A–C). The accelerated splenomegaly in imiquimod-treated Tlr9-/- mice was accompanied by increased levels of splenocyte TLR7 mRNA (figure 1D).
Supplemental material
Supplemental material

Deficiency of TLR9 increases splenomegaly associated with higher levels of TLR7 in imiquimod-induced autoimmunity. WT and Tlr9-/- mice were treated with topical imiquimod for 3 weeks. (A) Spleen weight. (B) Spleen weight/body weight. (C) Total number of splenocytes (WT-female-imiquimod n=14, Tlr9-/--female-imiquimod n=12, WT-male-imiquimod n=7, Tlr9-/--male-imiquimod n=8). (D) mRNA levels of TLR7 in the spleen (n=6/group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. TLR, toll-like receptor; WT, wild type.
While, as expected, imiquimod promotes higher levels of serum autoantibodies in the B6 WT mice, no significant differences in levels of total IgG, anti-dsDNA, antihistone and anti-RNP/SM autoantibodies were observed between imiquimod-treated Tlr9-/- and treated WT mice (figure 2A–D). However, different ANA patterns were observed between both groups. Specifically, 60% of the imiquimod-treated WT mice exhibited cytoplasmic speckled HEp-2 staining pattern, 30% exhibited both nuclear speckled and cytoplasmic speckled HEp-2 staining pattern and the rest 10% exhibited homogeneous HEp-2 staining pattern. In comparison, all the imiquimod-treated Tlr9-/- mice displayed both nuclear speckled and cytoplasmic speckled HEp-2 staining pattern, consistent with previous findings that speckled nuclear and cytoplasmic staining patterns are TLR7 dependent5 (figure 2E). These results indicate that TLR9 deficiency in imiquimod-induced autoimmunity promotes more severe splenomegaly associated with increased levels of TLR7 and a shift in ANA pattern.

Deficiency of TLR9 does not affect autoantibody levels but shifts autoantibody specificity. WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. Sera were collected and tested for (A) total IgG (WT-control n=5, Tlr9-/--control n=5, WT-imiquimod n=10, Tlr9-/--imiquimod n=10). (B) IgG anti-dsDNA autoantibodies (WT-control n=5, Tlr9-/--control n=4, WT-imiquimod n=10, Tlr9-/--imiquimod n=10). (C) IgG antihistone autoantibodies (WT-control n=5, Tlr9-/--control n=5, WT-imiquimod n=10, Tlr9-/--imiquimod n=10). (D) IgG anti-RNP/Sm autoantibodies (WT-control n=5, Tlr9-/--control n=4, WT-imiquimod n=10, Tlr9-/--imiquimod n=10). (E) Representative images of ANA. WT-control n=4, WT-imiquimod n=10, Tlr9-/--control n=4, Tlr9-/--imiquimod n=10. Bar, 75 µm. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. TLR, toll-like receptor; WT, wild type.
Imiquimod-treated Tlr9-/- mice display enhanced renal inflammation
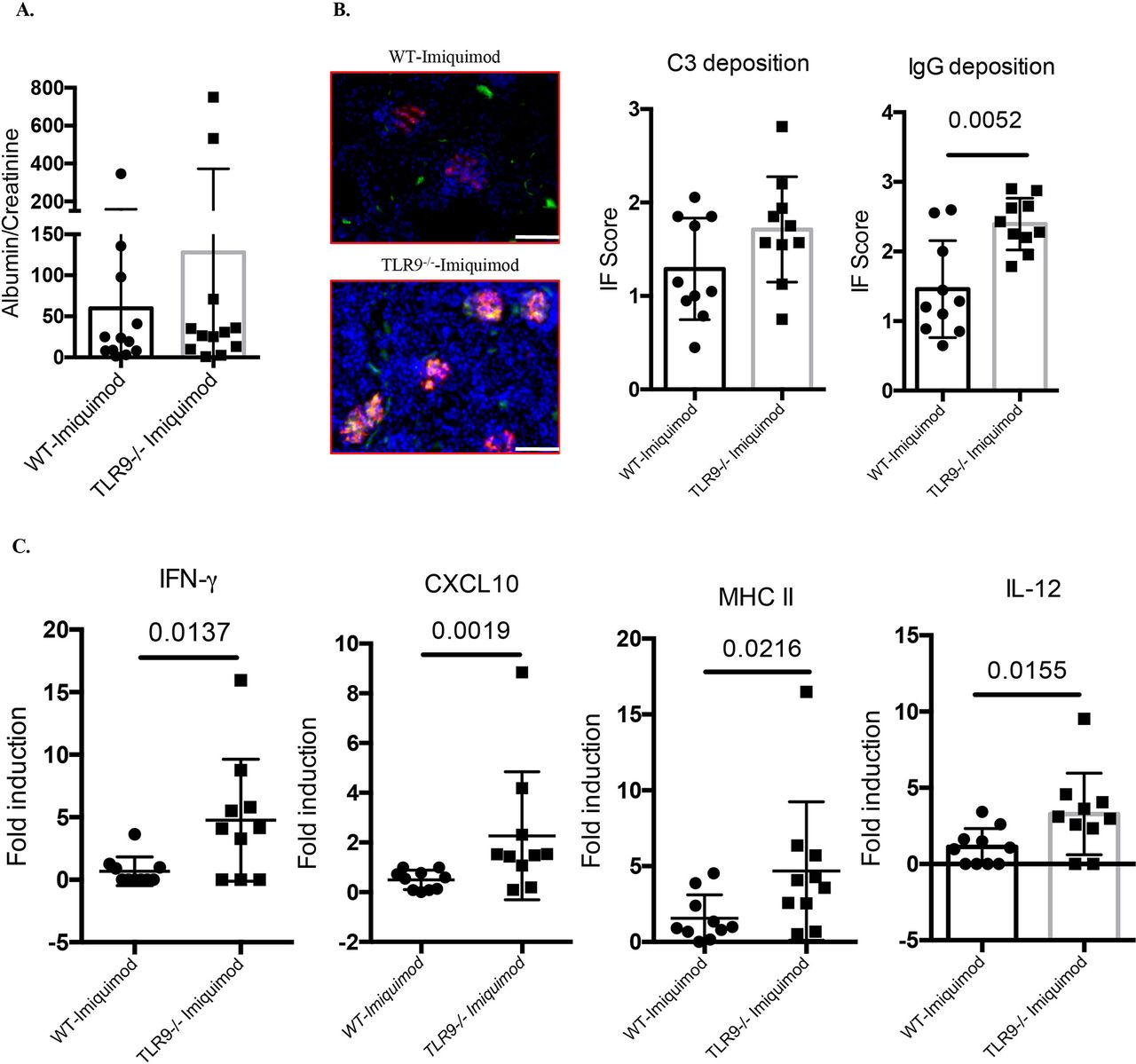
Imiquimod-associated autoimmunity is characterised by enhanced IC deposition in glomeruli. While we did not observe significant differences in the levels of mild proteinuria characteristic of this model (figure 3A), there was significantly more glomerular IgG deposition in imiquimod-treated Tlr9-/- mice compared with imiquimod-treated WT mice (figure 3B). In addition, gene expression profiling of kidney tissue revealed significantly higher levels of IFN-γ, CXCL10, MHC II and interleukin (IL)-12 p35 mRNAs in imiquimod-treated Tlr9-/- mice (figure 3C). Taken together, these data show that TLR9 deficiency in B6 imiquimod-induced autoimmunity leads to more severe kidney inflammation associated with increased IC deposition and increased mRNA levels of proinflammatory mediators.

Deficiency of TLR9 promotes more severe renal inflammation in imiquimod-induced autoimmunity. WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. (A) Urine was collected and tested for albumin and creatinine (n=12/group). (B) Representative images. Frozen kidney sections were stained for immune complex deposition, C3: green; IgG: red. Bar, 100 µm (n=10/group). (C) Gene expression of IFN-γ, CXCL10, MHC II and IL-12 p35 from kidney tissue (n=10/group). IFN, interferon; IL, interleukin; TLR, toll-like receptor; WT, wild type.
TLR9 deficiency exacerbates endothelial dysfunction in imiquimod-induced autoimmunity
Patients with SLE and genetically lupus-prone mouse models display significant impairments in endothelium-dependent vasorelaxation.17–22 We observed that mice exposed to imiquimod also display endothelial dysfunction, and tested whether this would be affected by TLR9 deletion. Indeed, imiquimod-treated Tlr9-/- mice exhibited more severe impairment in endothelium-dependent vasorelaxation (figure 4A). We previously showed that human and murine lupus endothelial progenitor cells (EPC) display decreased capacity to differentiate into mature endothelial cells.21 22 However, there were no differences in the differentiation capacity of EPCs when comparing untreated or imiquimod-treated WT and Tlr9-/- mice (online supplementary figure 1A), indicating that the vascular impairment in this model is not due to a defect of EPC function.

TLR9 deficiency leads to more severe impairment of endothelium-dependent vasorelaxation and promotes aberrant B cell phenotype in imiquimod-induced autoimmunity. WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. (A) Aortic rings were isolated from untreated and imiquimod-treated WT and TLR9-/- mice. Acetylcholine-dependent relaxation was determined and the reduction in maximal response (Emax) calculated. * is used to compare untreated WT versus imiquimod-treated WT, while # is used to compare untreated TLR9-/-with imiquimod-treated TLR9-/- mice. #P<0.05, ##P<0.01, *P<0.05, **P<0.01, ****P<0.001 (WT-control n=5, Tlr9-/--control n=6, WT-imiquimod n=5, Tlr9-/--imiquimod n=5). (B) WT and TLR9-/- mice were treated with imiquimod for 1 week. TLR7 expression was analysed in CD19+ B cells from spleens. MFI was calculated by the MFI=TLR7 MFI-isotype control MFI (WT-control n=3, Tlr9-/--control n=3, WT-imiquimod n=4, Tlr9-/--imiquimod n=4). (C) WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. Splenocytes were isolated and stained for CD45, CD19, CD40, CD80, CD86 and MHC II. Mean fluorescence intensity (MFI) was calculated (WT-imiquimod n=6, Tlr9-/--imiquimod n=6). (D) Splenocytes were isolated and stained for CD45, CD19, B220, FAS (CD95) and Peptide nucleic acid (PNA). (E) Splenocytes were isolated and stained for CD45, CD19 and CD138 (n=6/group). TLR, toll-like receptor; WT, wild type.
TLR9 deficiency promotes dysregulated B cell development and activation in imiquimod-induced autoimmunity
B cells are central to the pathogenesis of lupus by acting as antigen-presenting cells and autoantibody-producing cells.23 Imiquimod treatment led to significant elevation of TLR7 expression in splenic B cells on a per cell basis (figure 4B) and this was significantly enhanced in the imiquimod-treated Tlr9-/- mice (figure 4B). The expression of the costimulatory molecule CD40 was significantly elevated on B cells from imiquimod-treated Tlr9-/- mice (figure 4C). Imiquimod-treated Tlr9-/- mice also displayed a trend towards higher levels of B220+PNA+Fashi germinal centre B cells (figure 4D). In addition, the percentage of antibody-producing B cells (CD19lo/-CD138+ cells) in spleen was significantly higher in imiquimod-treated Tlr9-/- mice compared with treated WT mice (figure 4E).
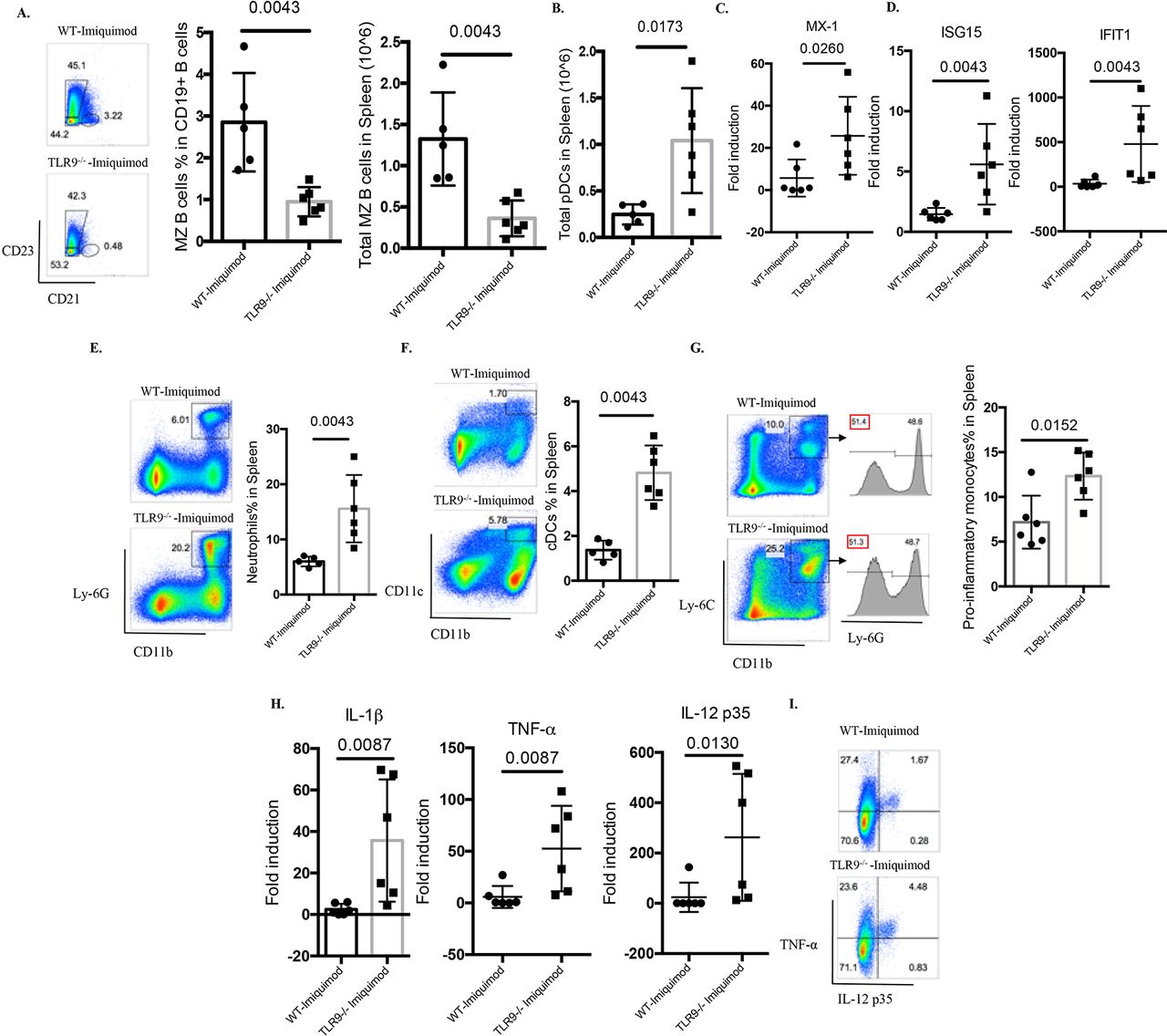
Abnormal B cell development has been observed in several lupus models, including those where TLR7 appears to play crucial roles.15 16 24 25 Indeed, Yaa mice and TLR7tg mice display hyperactive B cell phenotypes with marked loss of the marginal zone (MZ) B cell compartment.15 16 24 25 In the imiquimod-induced lupus model, previous evidence has found recapitulation of this abnormal B cell development phenotype, including the MZ B cell loss.14 While no differences were identified in the percentage of MZ B cells between untreated WT mice and untreated Tlr9-/- mice (online supplementary figure 1), imiquimod-treated Tlr9-/- mice display more prominent loss of MZ B cells compared with the WT mice (figure 5A), which may be due to DC-mediated enhanced activation (figure 5F), as suggested by Santiago-Raber et al.26 Taken together, these findings indicate that TLR9 deficiency leads to dysregulated B cell activation and B cell development in imiquimod-induced autoimmunity.

TLR9 deficiency promotes aberrant B cell activation and myeloid expansion in imiquimod-induced autoimmunity. WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. (A) Splenocytes were isolated and stained for CD45, CD19, CD21 and CD23 for MZ B cells. (B) Splenocytes were isolated and stained for CD45, CD11c, B220 and PDCA-1. Total pDC numbers were calculated. (C) mRNA levels of MX-1 in the spleen. (D) mRNA levels of ISG15 and IFIT1 in the spleen. (E) Splenocytes were isolated and stained for CD45, CD11b, Ly-6G for neutrophils. (F) Splenocytes were isolated and stained for CD45, CD11b, CD11c for CD11b+CD11c+ myeloid cells. (G) Splenocytes were isolated and stained for CD45, CD11b, Ly-6C and Ly-6G for proinflammatory monocytes. (H) mRNA levels of IL-1β, TNF-α and IL-12 p35 in the spleen. (I) Splenocytes were isolated and stained for CD45, CD11b and intracellularly stained for TNF-α and IL-12 p35 (n=6/group). cDC, conventional dendritic cell; IL, interleukin; MZ, marginal zone; pDC, plasmacytoid dendritic cell; TNF-α, tumour necrosis factor alpha; TLR, toll-like receptor; WT, wild type.
TLR9 deficiency increases type I IFN responses and expansion of myeloid cells in imiquimod-induced autoimmunity
Plasmacytoid DCs (pDC) are considered critical in initiation and maintenance of lupus through the synthesis of type I IFNs.27 28 pDC levels (figure 5B) and type I IFN-stimulated genes were significantly elevated in spleens of imiquimod-treated Tlr9-/- mice when compared with WT mice (online supplementary figure S4 and figure 5C,D). Splenic inflammatory monocytes were defined as CD11b+Ly-6C+Ly6G-, as described by Buechler et al.29 When quantifying splenic myeloid cells, neutrophils, CD11b+CD11c+ myeloid cells and inflammatory monocytes were all significantly increased in imiquimod-treated Tlr9-/- mice (figure 5E–G). Consistent with this myeloid expansion, the levels of splenic IL-1β, tumour necrosis factor alpha (TNF-α) and IL-12 were also significantly higher in these mice (figure 5H). By flow cytometry, we confirmed the enhanced production of TNF-α and IL-12 p35 by CD11b+ myeloid cells (figure 5I). These findings suggest that TLR9 deficiency leads to enhanced type I IFN synthesis and to aberrant expansion of myeloid subsets that contribute to the higher levels of proinflammatory mediators in imiquimod-induced autoimmunity.
Supplemental material
As we observed significantly increased splenomegaly and myeloid expansion in imiquimod-treated Tlr9-/- mice, we next evaluated whether deficiency of TLR9 affects bone marrow (BM) haematopoiesis (online supplementary figure S5A). Significantly increased frequencies of LSK cells, which are composed of long-term haematopoietic stem cells (HSC), short-term HSCs and multipotent progenitor cells, were observed in Tlr9-/- mice after 1 week of imiquimod treatment (online supplementary figure S5B,C). Interestingly, imiquimod-treated WT mice displayed significantly lower percentages of BM myeloid progenitor (MP) cells, common myeloid progenitors (CMP) and granulocyte myeloid progenitors (GMP) compared with untreated WT mice and imiquimod-treated Tlr9-/- mice after 1 week of imiquimod treatment, indicating that imiquimod may suppress MPs, GMPs and CMPs in WT mice, but this suppression is abrogated in Tlr9-/- mice (online supplementary figure S5B,C). Of note, at 3 weeks of imiquimod treatment, imiquimod-treated WT mice and imiquimod-treated Tlr9-/- mice displayed similar frequencies of LSKs, MPs, GMPs and CMPs (online supplementary figure S5D). Taken together, these findings suggest that TLR9 plays an important role in imiquimod-driven suppression of myelopoiesis.
Supplemental material
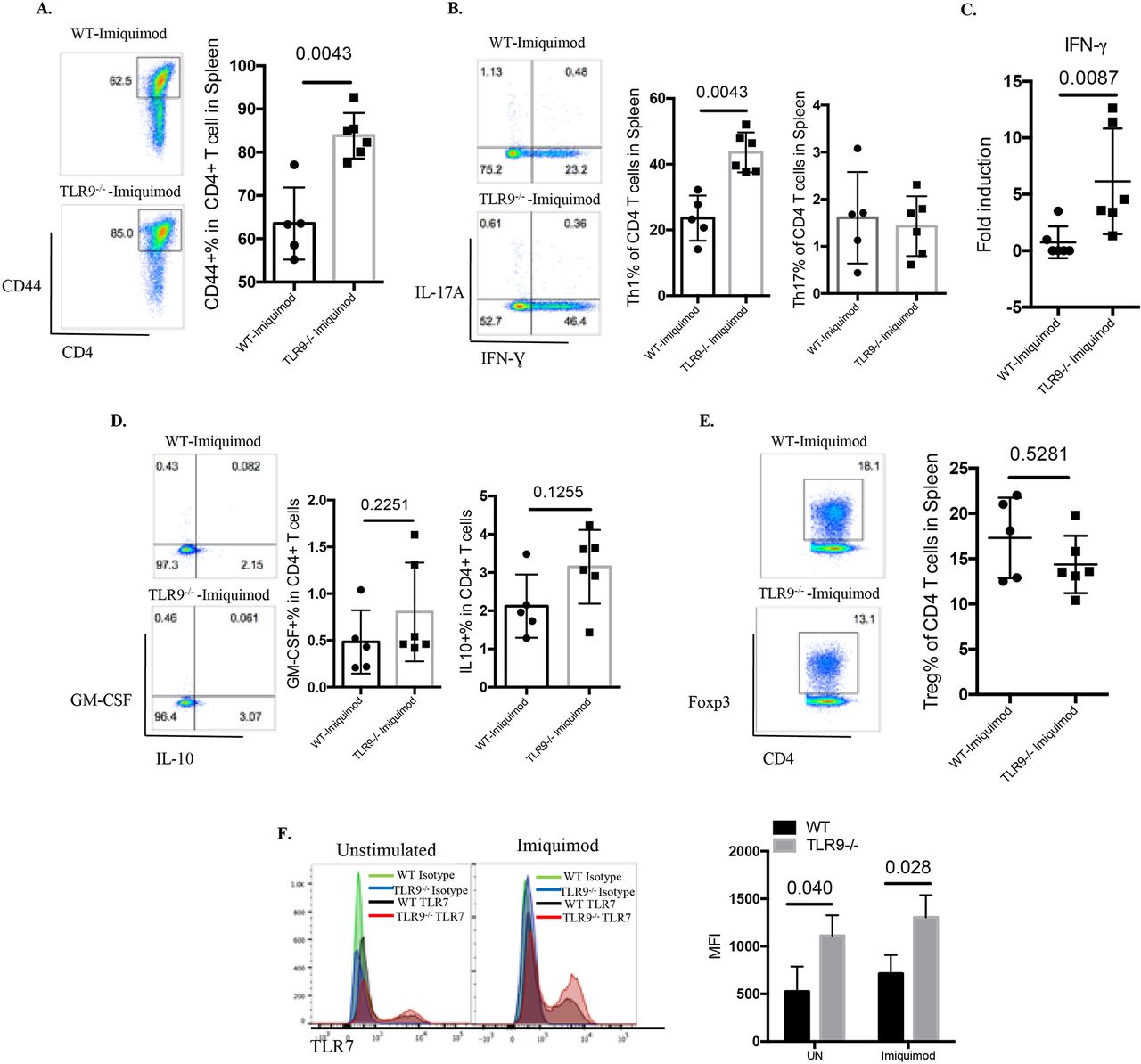
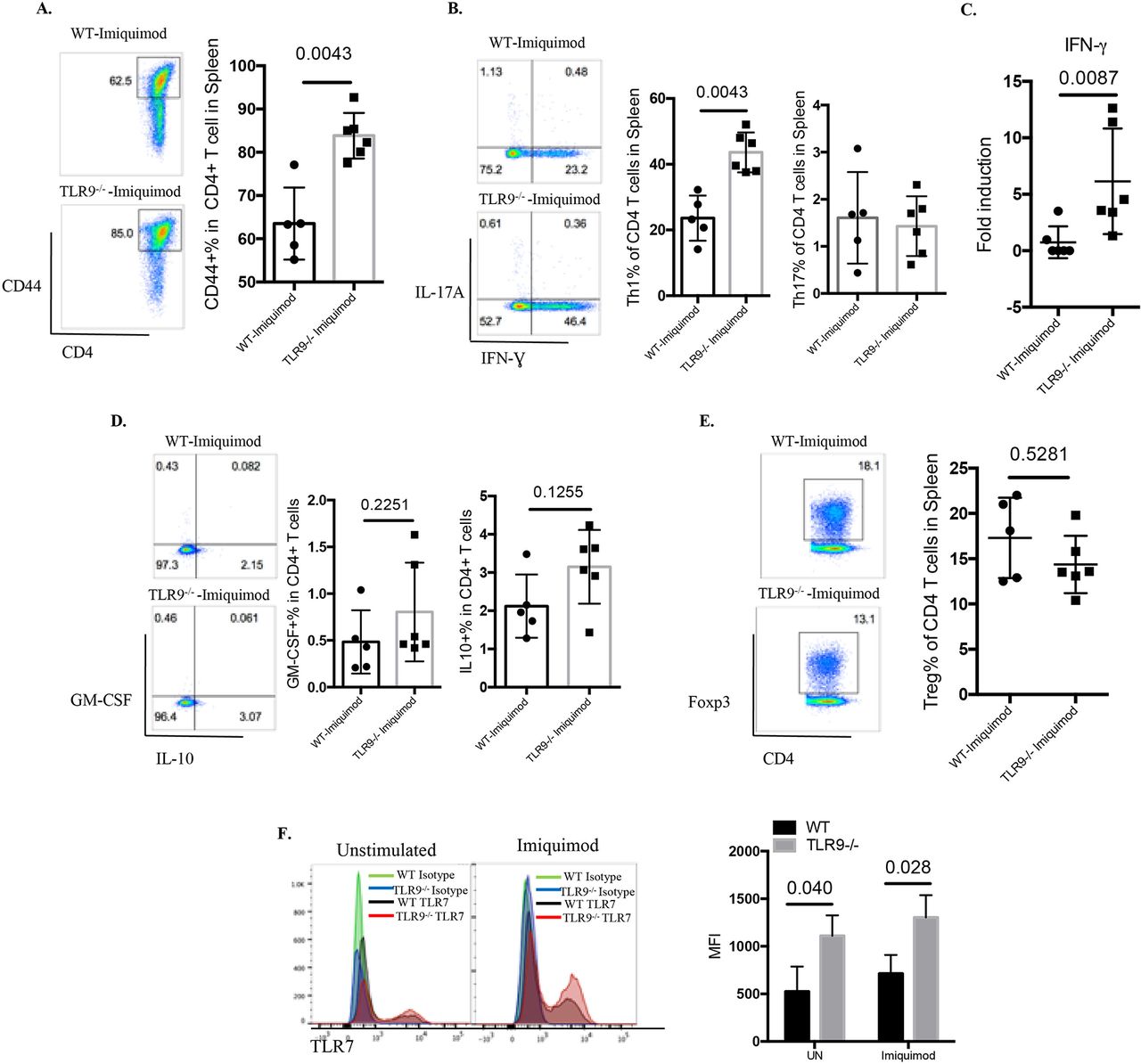
TLR9 deficiency promotes T cell activation and Th1 responses
T cells play major roles in SLE by amplifying autoimmune responses.30 31 T cells from imiquimod-treated Tlr9-/- mice were more activated than those from imiquimod-treated WT mice (figure 6A). Further analysis of the CD4+ T cell subsets revealed a significant elevation of IFN-γ-producing Th1 cells with minimal changes in the Th17 compartment in imiquimod-treated Tlr9-/- mice compared with imiquimod-treated WT mice (figure 6B). This was consistent with increases in the Th1 polarising cytokine IL-12 in imiquimod-treated Tlr9-/-mice (figure 5H) (figure 5K). Gene expression analysis also confirmed the elevated levels of splenic IFN-γ mRNA in imiquimod-treated Tlr9-/- mice (figure 6C). No significant differences in the levels of granulocyte-macrophage colony-stimulating factor-producing CD4+ T cells, IL-10-producing CD4+ T cells or FoxP3+ regulatory T cells were observed between imiquimod-treated WT mice and imiquimod-treated Tlr9-/- mice (figure 6D,E). These findings suggest that aberrant T cell activation and preferential polarisation to Th1 cells may contribute to the increased levels of IFN-γ in the kidney and to disease severity in imiquimod-treated Tlr9-/- mice.
![[lupus-2018-000259-SP2.jpg]](https://lupus.bmj.com/content/lupusscimed/5/1/e000259/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
![[lupus-2018-000259-SP3.jpg]](https://lupus.bmj.com/content/lupusscimed/5/1/e000259/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![[lupus-2018-000259-SP5.jpg]](https://lupus.bmj.com/content/lupusscimed/5/1/e000259/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
![[lupus-2018-000259-SP6.jpg]](https://lupus.bmj.com/content/lupusscimed/5/1/e000259/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
TLR9 deficiency promotes aberrant T cell and myeloid dendritic cell (DC) phenotype in imiquimod-induced autoimmunity. WT and Tlr9-/- mice were treated with imiquimod for 3 weeks. (A) Splenocytes were isolated and stained for CD45, CD4 and CD44. (B) Splenocytes were isolated and stained for CD45, CD4 and intracellularly stained for IFN-γ and IL-17A. (C) mRNA levels of IFN-γ in the spleen. (D) Splenocytes were isolated and stained for CD45, CD4 and intracellularly stained for GM-CSF and IL-10. (E) Splenocytes were isolated and stained for CD45, CD4 and intracellularly stained for FoxP3 (n=6/group). (F) Bone marrow-derived DCs (BMDC) from WT and Tlr9-/- mice were stimulated with medium (UN) and imiquimod (1 µg/mL) for 24 hours. TLR7 expression was determined by flow cytometry on CD11c+ DCs (n=4). GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; MFI, mean fluorescence intensity; TLR, toll-like receptor; WT, wild type; UN, unstimulated.
TLR9 deficiency promotes enhanced DC responses to imiquimod
As we observed a predominant Th1 response, we next assessed the phenotype of BM-derived DCs (BMDC) in response to imiquimod and IFN-γ. Imiquimod stimulation strongly induced IL-12 gene expression, and imiquimod plus IFN-γ exerted additive and/or synergistic effects on IL-12 mRNA levels (online supplementary figure S5E). Tlr9-/- BMDCs showed significantly higher levels of IL-12 mRNA in response to imiquimod or imiquimod plus IFN-γ (online supplementary figure S5E). Imiquimod and imiquimod plus IFN-γ also induced a moderate increase in TLR7 mRNA expression that was significantly higher in TLR9-/- BMDCs (online supplementary figure S5F). Flow cytometry analysis demonstrated significantly higher TLR7 expression on a per cell basis in Tlr9-/- BMDCs in unstimulated conditions or after imiquimod (figure 6F). Overall, these results indicate that lack of TLR9 in the context of TLR7 stimulation induces an overactivated myeloid DC phenotype.
Discussion
In this study, we used an induced lupus model that uses epicutaneous application of TLR7 agonists to investigate the role of a TLR7/TLR9 imbalance in the development of systemic autoimmunity and lupus-associated vasculopathy, independent of lupus susceptibility genes. This inducible model has clinical relevance as there is evidence that imiquimod can induce cutaneous lupus with autoantibody production in humans.32–34 Using this model, we demonstrate that TLR9 deficiency results in an accelerated autoimmunity and inflammatory phenotype that develops within 3 weeks of imiquimod treatment. Although the exacerbation of lupus in Tlr9-/- mice has been reported in several studies, the exact mechanisms remain unclear. In our model, increased disease severity in Tlr9-/- mice involves complex interactions among multiple cell lineages. Imiquimod treatment leads to significantly higher levels of TLR7 in Tlr9-/- mice, which results in profound expansion and activation of myeloid cells with enhancement of the proinflammatory phenotype, in association with lymphocyte activation and preferential differentiation to IFN-γ-producing Th1 cells. The IFN-γ-producing T cells are recruited to the kidney and may contribute to exacerbated renal pathology. Furthermore, and as a novel finding, the lack of TLR9 also exacerbates vasomotor dysfunction triggered by TLR7 agonists, an important phenomenon considering that patients with lupus suffer from enhanced vasculopathy and accelerated vascular risk.17 18 These findings underscore the protective role of TLR9 in TLR7-driven autoimmunity, further reinforcing the importance of tightly manipulating TLRs in putative therapeutic strategies.
Yokogawa et al have shown that topical administration of imiquimod led to pDC infiltration and accumulation in the skin where imiquimod was applied, and depletion of pDCs ameliorates the disease.14 In fact, the lupus-like phenotype was only observed through epicutaneous application of imiquimod, but not through intraperitoneal administration of imiquimod, indicating an essential role for pDCs in the initiation of systemic autoimmunity.14 In our study, we found that the levels of pDCs and IFN-response genes (MX-1, ISG15 and IFIT1) were significantly elevated in imiquimod-treated Tlr9-/- mice, which may contribute to the accelerated expansion and activation of both innate and adaptive immune arms in imiquimod-treated Tlr9-/- mice.
One important factor driving the severe splenomegaly in imiquimod-treated Tlr9-/- mice was the expansion of the myeloid compartment, most strikingly the neutrophils and to a lesser extent, the proinflammatory monocytes and myeloid DCs. In fact, TLR7tg mice also display severe splenomegaly characterised by peripheral myeloid expansion, characterised by expansion of myeloid compartment, including CD11b+CD11c+ myeloid cells, inflammatory monocytes and neutrophils.15 29 A recent study using Tlr9-deficient Fas-sufficient MRL/+ lupus-prone mice or pristane-induced lupus on Tlr9-deficient BALB/c background also reported a similar expansion of the myeloid compartment,11 35 similar to the Yaa mice and TLR7tg mice.29 36 These findings suggest that increases in gene dosage of Tlr7 may drive the myeloid expansion, particularly when no compensation by TLR9 exists.
The number of germinal center (GC) B cells was also significantly increased in imiquimod-treated Tlr9-/- mice, similar to the B cell phenotype described in the TLR7tg mice.37 Furthermore, B cells from imiquimod-treated Tlr9-/- mice displayed higher TLR7 levels, consistent with previous observations demonstrating that excess TLR7 promotes GC B cell and plasmablast development38 and MZ B cell loss, and that deletion of TLR9 further increases GC B cells and CD138+ plasma cells/plasmablasts.39 Our observations are then consistent with the hypothesis that B cell-intrinsic TLR9 signals function to limit B cell activation.37
Interestingly, TLR9 has been shown to compete with TLR7 for Unc93B1-dependent trafficking and predominates over TLR7, thus modulating the expression of TLR7.40 We found that imiquimod significantly increases TLR7 levels both in vivo and in vitro (figures 4A and 6F, and online supplementary figure S5F). Thus, TLR9 deficiency further enhanced imiquimod-induced TLR7 expression in imiquimod-induced lupus model. Interestingly, Fukui et al described that mice harbouring a D34A mutation in Unc93B1, which skews the TLR7 and TLR9 balance to TLR7, showed TLR7-dependent systemic lethal inflammation, characterised by huge splenomegaly, expansion of myeloid cells, loss of MZ B cells, autoantibody production and enhanced T cell differentiation,40 which is similar to what we observed in our model.
In the present study, we did not observe significantly differences in terms of levels of anti-dsDNA autoantibodies between imiquimod-treated WT mice and imiquimod-treated Tlr9-/- mice. Interestingly, conflicting data have been shown regarding the role of tlr9 in generating anti-dsDNA autoantibodies from different murine lupus models. Christensen et al did not find differences in anti-dsDNA autoantibodies between WT mice and Tlr9-/- mice in lupus-prone MRL/Mpj/lpr/lpr background,4 while other models reported either increased 41 or decreased levels of anti-dsDNA autoantibodies,42 indicating the other factors may also contribute to generation of these type of Abs. Thus, the role of tlr9 in generation anti-dsDNA autoantibodies may be model dependent, further investigations are strongly needed.
In imiquimod-treated Tlr9-/- mice, we also found significantly higher levels of Th1 cells compared with imiquimod-treated WT mice. This may be due both to the higher levels of IL-12 synthesised by the myeloid compartment and to higher levels of CD40 and CD80 on B cells, known to be critical in driving T cell activation differentiation.43 44
Significantly higher levels of renal IC deposition were detected in imiquimod-treated Tlr9-/- mice compared with imiquimod-treated WT mice. Similar differences were described in pristane-treated Tlr9-/- mice on BALB/c background.11 The exact mechanisms leading to this phenotype remain to be further elucidated. As levels of autoantibodies were not appreciably different between imiquimod-treated Tlr9-/- mice and treated WT mice, changes in IC deposition may be at least partially attributable to a higher level of infiltration of inflammatory cells and perhaps skewing in inflammatory cytokine profiles. IFN-γ-producing T cells promote renal immune deposits without promoting autoantibody production.45 Furthermore, deletion of IL-12 in MRL/faslpr mice can decrease systemic and intrarenal IFN-γ-secreting cells with dramatic reductions in renal pathology without affecting autoantibodies.46
Importantly, we demonstrate that the imiquimod-induced lupus mice have significant evidence of endothelial injury. This is potentially clinically relevant as endothelial dysfunction is highly prevalent in lupus, predating atherosclerotic clinical disease.17 We describe, for the first time to our knowledge, that TLR9 is protective in lupus-associated vascular damage. While the mechanisms leading to the exacerbated vasculopathy in imiquimod-treated Tlr9-/- mice are yet unclear and do not appear to be dependent on EPC function, the profound inflammatory environment is likely a major contributing factor. Indeed, IFN-γ, TNF-α and type I IFN,47–49 which are potentially involved in vascular dysfunction, were significantly elevated in imiquimod-treated Tlr9-/- mice. Of interest, untreated Tlr9-/- mice also displayed worse endothelium-dependent vasorelaxation compared with untreated WT mice. Additional studies are required to determine whether TLR9 may exert an intrinsic protective role in maintaining endothelial homeostasis. While the role of TLR9 in atherosclerosis is controversial, there is some evidence indicating a protective role for this TLR in the development of atherosclerosis in models like the apolipoprotein E-deficient mice.50
The original description of the imiquimod model of lupus autoimmunity did not find the same increased response to imiquimod in TLR9-null mice compared with WT mice when the BALB/c background was studied.14 The discrepancy may be due to the differences in mice genetic background and future studies should explore if these phenotypes can be recapitulated in other non-autoimmune prone strains and whether factors such as variations in microbiome composition could modulate the differential responses.
While conflicting results from multiple ethnicities regarding the associations of Tlr9 polymorphisms with increased lupus risks have been shown,51–54 there is some evidence that in certain patient populations, polymorphisms in Tlr9, which result in downregulation of TLR9 expression, are closely associated with increased risk of SLE.51 A reported genetic association between SLE and single nucleotide polymorphisms within Tlr9 genes also found that in these individuals mRNA TLR7 levels were significantly elevated compared with healthy controls.54 As such, these individuals may be at higher risk to develop autoimmunity upon exposure to endogenous or exogenous TLR7 agonists.
Better and more targeted therapeutics for SLE are clearly needed. Our study supports the protective role of TLR9 in TLR7-driven autoimmunity, further providing critical insights into the importance of tightly manipulating TLRs in putative therapeutic strategies, and provides an additional model of induced accelerated autoimmunity and vascular damage.
Supplemental material
![[lupus-2018-000259-SP4.jpg]](https://lupus.bmj.com/content/lupusscimed/5/1/e000259/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
Acknowledgments
The authors thank Dr Daniela Verthely (FDA) for providing initial breeding pairs of Tlr9-/- mice and Dr Shizuo Akira (Osaka University, Japan) for the permission to use the Tlr9-/- mice. The authors thank the NIAMS Office of Science and Technology for technical support including the Flow Cytometry Group and the NIAMS Laboratory Animal Care and Use Section. The authors also thank Dr Zu-Xi Yu (Pathology Core, NHLBI/NIH) for technical assistance.
References
Footnotes
Contributors YL and MJK conceived the study. YL, NLS and CCR performed experiments and statistical analysis. YL and MJK wrote the manuscript.
Funding This work was supported by the Intramural Research Program at NIAMS/NIH (ZIA AR041199).
Competing interests None declared.
Patient consent Not required.
Ethics approval NIAMS-approved animal study protocol (No A016-05-26).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.