





Abstract
Several studies have found that smoking cigarettes is a risk factor for systemic lupus erythematosus (SLE). To examine this issue in a mouse model, we subjected pre-autoimmune MRL-lpr/lpr mice for 4 weeks to cigarette smoke to provide standardized smoke effluents equivalent to moderate or to heavy smoking habits for people. The spontaneous production of IgG anti-chromatin but not IgM anti-chromatin, anti-denatured DNA, or rheumatoid factor antibodies was lower in mice exposed to 250 mg/m3 particulates from mainstream smoke, and this suppression of autoimmunity was sustained for 8 weeks (p < 0.02). In contrast to control mice anti-chromatin activity in smoke-exposed mice began to increase in 16-week-old mice, reaching levels at 6 months that were two- to three-fold higher than controls for IgG (p < 0.03) and 10-fold higher for IgM (p < 0.001). There was no significant effect on total IgG or IgM. In newly diagnosed SLE patients, smoking was negatively correlated with IgG anti-DNA antibodies (p < 0.03). However, of nine patients who discontinued smoking prior to diagnosis, eight had elevated IgG anti-DNA compared to 29/79 never smokers and 9/31 smokers (p < 0.01 compared to former smokers). Inhaled cigarette smoke appears to have a long-lasting immunsuppressive effect on T-cell-dependent autoimmune responses, although autoantibodies increase to supra-elevated levels after the suppressive effect has abated.
Environmental factors are widely believed to influence lupus-like syndromes, although the heterogeneity of diseases such as systemic lupus erythematosus (SLE) in patients with markedly different genetic backgrounds and the general ignorance regarding etiopathogenesis make a role for external factors difficult to prove. Cigarette smoke remains one of the greatest sources of environmental exposure to toxic chemicals and reactive molecular species (Selgrade et al., 1999), and the majority of studies have shown that smoking cigarettes is a risk factor for SLE (Costenbader et al., 2004). However, odds ratios varied widely among studies from a high of 6.7 (Ghaussy et al., 2001) to a negative association of 0.9 (Reidenberg et al., 1993), suggesting that differences in patient populations and study designs as well as unknown factors profoundly affect the association between smoking and SLE. Identifying these variables and understanding mechanisms for putative aggravating effects of cigarette smoking on SLE may be relevant to other diseases as well.
The positive association between cigarette smoking and SLE is commonly interpreted as indicating that smoking increases the risk for the disease or enhances disease activity. However, distinguishing cause and effect—does smoking precipitate and/or worsen SLE, or does affliction with SLE promote smoking?—cannot be readily discriminated by these observations. It is possible that SLE patients may be predisposed to smoking cigarettes because its psychological and/or physiological effects are palliative in systemic autoimmune disease, especially in lupus patients with a central nervous system disease component.
Proposed explanations for the association between cigarette smoke and autoimmunity include release and alteration of autoantigens due to toxic agents in inhaled smoke, mutation of immunoregulatory genes, and increasing susceptibility to putative infectious triggers of autoimmunity due to immunosuppressive qualities of smoking (reviewed in Holt, 1987; Sopori, 2002). However, while there is support for each of these mechanisms from studies unrelated to smoking exposure, none has been firmly established as important in the etiology of autoimmunity. The difficulty in undertaking mechanistic studies in humans due to subject heterogeneity, ethical limitations to experimental protocols, and psychosocial complications necessitates the use of animal models to explore these issues. We hypothesized that, if smoking were to have a fundamental, causative relationship with SLE, then inhalation of cigarette smoke would accelerate or exacerbate disease in animals predisposed to this syndrome.
There are no prior studies on the effects of inhaled cigarette smoke on the immune status of autoimmune-prone mice. Therefore, we exposed MRL-lpr/lpr mice to cigarette smoke in a state-of-the-art toxicological exposure facility where the dose and duration of exposure could be controlled. MRL-lpr/lpr mice are considered a model of global systemic autoimmunity, with features of SLE, rheumatoid arthritis, and Sjögrens syndrome (Theofilopoulos and Dixon, 1987). Signs of B-cell activation begin at approximately 6 weeks of age, and half the mice will die of glomerulonephritis by the age of 6 months (Hahn, 2002). In order to test for acceleration of autoimmunity, mice were exposed to cigarette smoke soon after weaning, prior to expression of overt signs of autoimmunity, and then followed for autoantibody expression until death.
Interpretation of prior studies on the association of cigarette smoking with SLE may be complicated by patients' use of medications that can alter disease. Also, the pharmacokinetics of many drugs can be affected by simultaneous cigarette smoke intake (Miller, 1990). For these reasons we analyzed the immune status of SLE patients stratified according to smoking history at the time of diagnosis before treatment was initiated in order to afford better comparison to the mouse data. While our results suggest that cigarette smoke suppresses lupus, SLE patients who continue to smoke cigarettes may have worse disease, in part because of undertreatment with anti-inflammatory or immunosuppressive medications due to smoking-induced reduction in effective drug levels.
MATERIALS AND METHODS
Cigarette smoke exposure protocol.
Four-week-old female MRL/Mp-lpr/lpr mice (Jackson Laboratories, Bar Harbor, ME) were individually caged without bedding in 2-m3 wire-bottom stainless steel exposure chambers ventilated with clean air at 12 ± 2 air-changes/h. After 2 weeks conditioning, eight animals were randomly assigned by weight to one of three experimental groups and identified by unique ear tags. Tap water was available via automatic valves ad libitum, and pelleted ration (Harlan Teklad Certified Rodent Diet, Indianapolis, IN) was available after exposure hours. Mice assigned to receive cigarette smoke were exposed for 4 weeks, 6 h/day, 5 days/week. All exposed mice were acclimated to 100 mg total particle mass (TPM)/m3 mainstream cigarette smoke for 1 week. Mice were then divided into two groups, receiving smoke doses of either 250 mg or 100 mg TPM/m3. The control mice were exposed to HEPA-filtered air under otherwise identical conditions. Smoke was produced from a modified commercial machine (Type 1300, AMESA, Geneva, Switzerland) from filtered 2R4F research cigarettes (Tobacco Health Research Institute, Lexington, KY) using a peristaltic pump that generated twice/min a 70-ml puff during 2 s. Smoke was diluted with HEPA-filtered air. Exposure TPM concentrations were determined gravimetrically by collecting air samples 3×/day for 2 h from the mid-point of each chamber onto a 25-mm glass fiber filter (Type A/E, Gelman, Ann Arbor, MI). Actual exposure concentrations were within 10% of targeted values on each day of the study.
After the 4-week exposure period, the cages were moved to another room. Biological fluids were collected every 2–4 weeks for about 4 more months. Blood was obtained from the intraorbital sinus and immediately processed for serum storage. Urine from individual mice was collected into a specimen cup during spontaneous grasp response in the morning prior to blood draw. All eight mice/group were alive at 16 weeks (6 weeks after the end of the smoke exposure period), and 5–6 mice/group were alive at 25 weeks of age. The protocol was halted 1 month later when <50% of the mice were alive. There was no significant difference in the mortality of mice among the three exposure groups based on average time to death or number of mice that died (monitored daily). This protocol was approved by both the University of New Mexico and the Lovelace Respiratory Research Institute animal care and use committees.
Autoantibody determinations.
Autoantibodies in mice to chromatin, denatured DNA, and IgG were quantified by ELISA as previously described (Burlingame and Rubin, 1990, 2002). Briefly, Immulon 2HB microtiter plates (Dynex Laboratories, Inc., Alexandria, VA) were coated with antigen at 2.5 μg/ml. For the anti-chromatin assays, H1-stripped chromatin was used as the solid-phase antigen. S1-nuclease-treated DNA was used in the anti-native DNA assay, and DNA was heated for 10 min and quick cooled for the denatured (d) DNA. For the IgM rheumatoid factor (anti-IgG) assay, IgG2a myeloma protein (Bethyl Laboratories, Inc., Montgomery, TX) was used as the antigen because prior studies demonstrated this isotype as the dominant target for IgM rheumatoid factors in MRL-lpr/lpr mice (Rubin et al., 1984). Sera were diluted 1:200, and the bound antibodies were detected with peroxidase-conjugated anti-mouse IgG or IgM (Caltag, San Francisco, CA) and 2,2′ azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) secondary substrate. Optical densities (OD) beyond the range of direct measurement at 1 h in the ELISA were extrapolated from OD at earlier time-points as described (Burlingame and Rubin, 1990). Positive and negative control sera were included in each assay, and values determined in different assays were normalized by multiplying by the ratio of the reactivity of the positive control sera tested in both assays.
Proteinuria determination.
Protein was measured immediately after urine collection using Chemstrip 2 GP urine test strips (Roche Diagnostics Corp., Indianapolis, IN) according to the manufacturer's instructions.
Immunoglobulin determinations.
Total IgG and IgM levels were determined by capture ELISA as previously described (Bell et al., 1992; Burlingame and Rubin, 2002). Briefly, for IgG determination an anti-mouse kappa chain antibody (Caltag) was coated on Immulon 2HB plates, which were then incubated with mouse sera generally diluted 1:200,000. Bound IgG was detected with anti-mouse IgG coupled to horseradish-peroxidase followed by ABTS. For IgM determination, anti-mouse IgM (Caltag) was used as the capture antibody, sera were generally diluted 1:5000, and bound IgM detected with anti-mouse IgM coupled to horseradish-peroxidase. Standard curves were generated simultaneously using serial dilutions of polyclonal mouse IgG (Zymed Laboratories, Inc., South San Francisco, CA) in the range of 5–125 ng/ml or polyclonal mouse IgM (Litton Bionetics, Kensington, MD) in the range of 15–500 ng/ml. These values were used to fit a binomial expression in which OD = ax2 + bx + c, and Ig levels in serum were determined by converting OD to ng/ml (x-value) using the quadratic formula. Each sample was assayed at least twice, and for samples that were beyond the range of the standard curve, determinations were repeated at higher dilutions.
Systemic lupus erythematosus patients.
SLE subjects participating in the University of New Mexico Lupus Cohort, which is a database consisting of all SLE patients encountered on both the inpatient and outpatient services, were consecutively enrolled in the present study. The study consisted of a search of the database for SLE subjects with early SLE and without therapy with antimalarials, corticosteroids, or immunosuppressive agents. Inclusion criteria were (1) female gender, (2) disease duration based on date of diagnosis of 1 year or less (defined as early SLE), and (3) no therapy with antimalarials, corticosteroids, or immunosuppressives.
The diagnosis of SLE was established in each subject using the American Rheumatism Association 1982 (Tan et al., 1982) and American College of Rheumatology (ACR) 1997 revised criteria for SLE (Hochberg, 1997), and these criteria were compared between the smokers and nonsmokers. The diagnosis of SLE was confirmed by a rheumatologist after an in-depth face-to-face interview, medical history, physical examination, chart-review, and appropriate laboratory testing. SLE disease activity was determined with the SLE Disease Activity Index (SLEDAI) (Bombardier et al., 1992; Hawker et al., 1993), and SLE disease severity (damage index) was measured with Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index (SLICC/ACRDI) (Gladman et al., 1996), both of which are categorical scales that include clinical and laboratory data from individual patients. The Neuro-SLEDAI is composed of the neurologic components of SLEDAI—seizure, psychosis, organic brain syndrome, visual disturbance, cranial neuropathy, lupus headache, stroke syndrome—each of which is given an arbitrary score ranging from 0 to 8 and then summed (Bombardier et al., 1992; Ghaussy et al., 2001, 2003; Hawker et al., 1993; Sibbitt et al., 2003). The Neuro-SLICC measures only the neurologic components of SLICC (retinal or optic atrophy, cognitive disorder or psychosis, seizures, stroke syndrome, cranial neuropathy, transverse myelitis) (Gladman et al., 1996; Ghaussy et al., 2001, 2003; Sibbitt et al., 2003).
Autoantibody determinations were performed as follows. Anti-DNA levels were quantified by titration up to 1:640 using a Crithidia luciliae immunofluorescence assay (Aarden et al., 1975) standardized against international standards (Feltkamp et al., 1988). Anti-phospholipid antibodies of IgG, IgM, and IgA isotypes were measured using ELISA (Merkel et al., 1999) (APhL ELISA Kit, Louisville APL Diagnostics, Inc., Louisville, KY) using appropriate isotype calibrators. Rheumatoid factor was measured by nephelometry (Collins et al., 1990) and expressed as international units.
Cigarette smoking was defined as (1) never smoked, (2) current (smoking within 2 months of interview), and (3) previous (no cigarettes for greater than 2 months). Cigarette smoking intensity was defined in packs/day or fraction thereof for both current and previous smokers. A person who smoked less than 0.25 pack/day was considered a nonsmoker. Informed consent of each patient was obtained, and this study was approved by the institutional review board.
Statistical analyses.
To evaluate the autoantibody data derived from serial samples of mouse serum, a variation of a standard repeated measures model was used to compare the mean anti-chromatin levels at the seven post-exposure dates across treatment groups. A standard split-plot repeated measures analysis was inappropriate because the variances of the autoantibody levels increased with time, and the correlation between levels depended on time. The initial model included a random effect for each mouse and group-specific variance functions that allowed for increasing variability with time. Alternative variance and correlation models were considered, with no appreciable differences in conclusions. All analyses were performed using the mixed model procedure in SAS (Release 8.02, SAS Institute Inc, Cary, NC, 1999).
The data from the SLE patients was analyzed in several ways. For binary data (presence or absence of abnormality) p-values were computed using Fisher's exact test. Laboratory values on continuous scales were nonnormally distributed, so p-values were based on the nonparametric Wilcoxon-Mann-Whitney test. Logistic regression analysis was used in some cases as noted.
RESULTS
Effect of Cigarette Smoke on Development of Autoimmunity in MRL-lpr/lpr Mice
Groups of eight MRL-lpr/lpr mice at 6 weeks of age, prior to appearance of serum autoantibodies, were individually exposed to smoke produced by a cigarette smoking machine. The three exposure levels of 0, 100, and 250 mg TPM/m3 atmospheric smoke were maintained for 30 h/week for 4 weeks. The exposure concentration of 100 mg TPM/m3 used in this study was reported to produce a plasma cotinine (a stable nicotine metabolite) level of 120 ng/ml in A/J mice (Witschi et al., 2002), equivalent to the steady-state cotinine level while a person is smoking 0.50–0.75 packs/day (Peacock et al., 1998); systemic cotinine levels produced by 250 mg TPM/m3 have not been measured in mice, but based on calculations of lung deposits of smoke TPM in rats exposed to this level of cigarette smoke, are estimated to simulate humans smoking 2–3 packs/day (Finch et al., 1998; Mauderly et al., 2004). However, since the mice were exposed to cigarette smoke for only 6 h/day and 5 days/week, plasma levels of smoke components while exposure was suspended would be substantially lower than in human smokers, who would normally smoke every day and throughout the day.
Blood and urine were periodically sampled while the mice were exposed to cigarette smoke and over the subsequent 4 months and tested for signs of autoimmunity. Figure 1 shows the mean IgG anti-chromatin antibodies in mice of each group from the preexposure period to the time when approximately 50% of the mice had died at 6 months of age and the experiment was discontinued. Control MRL-lpr/lpr mice displayed a large increase in IgG anti-chromatin activity over a 12-week period, and these antibodies began to decline when the mice were about 16 weeks old. Mice exposed to 250 mg/m3 cigarette smoke showed a delayed appearance of IgG anti-chromatin antibodies, resulting in significantly lower levels than the control group when mice were 18 weeks old (p < 0.04). Also, in contrast to the controls, IgG anti-chromatin activity in mice exposed to both 100 mg and 250 mg smoke/m3 continued to increase for the next 8 weeks, reaching levels in 25–30 week old mice that were two- to three-fold higher than the controls. Statistical analysis at individual time points demonstrated significantly elevated IgG anti-chromatin activity at 23.4 weeks in the 100 mg/m3 smoke-exposed mice (p < 0.023) and at 23.4 and 25.6 weeks in 250 mg/m3 smoke-exposed mice (p < 0.04), 4 months after cessation of the smoke exposure protocol.
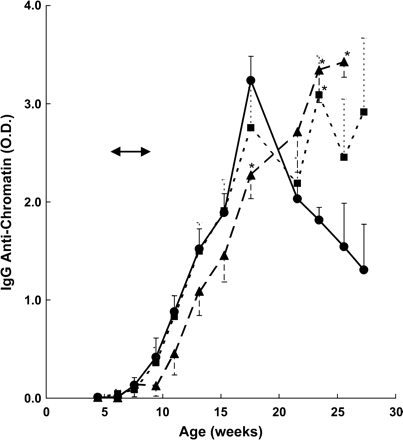
IgG anti-chromatin activity in MRL-lpr/lpr mice exposed to cigarette smoke. IgG serum autoantibodies were measured by ELISA. Cigarette smoke exposure period is marked by arrows and refers to a dose of 0 (• —— •), 100 (▪- - - - - ▪), or 250 (▴ – – – ▴) mg TPM/m3. Variances are SE, and asterisks indicate significant differences compared to control at individual time points shown. All eight mice/group were alive at 16 weeks, and five or six mice/group were alive at 23 weeks. The protocol was halted at 27 weeks when three, four, and one mice survived at 0, 100, and 250 mg TPM/m3, respectively.
MRL-lpr/lpr mice in the control group displayed a more limited production of IgM anti-chromatin antibodies compared to autoantibodies of the IgG isotype, approaching an activity maximum at about 18 weeks, followed by a small decline (Fig. 2). In the smoke-exposed mice the kinetics of IgM anti-chromatin antibody appearance was similar up to 18 weeks, after which these antibodies continued to increase, reaching two- to three-fold higher levels in the 100 mg smoke-exposed animals and >10-fold increase in animals exposed to 250 mg smoke/m3. A test of no difference in mean levels among the treatment groups considering all time points simultaneously was highly significant (p < 0.001); pairwise comparison of groups showed that the mean IgM anti-chromatin levels for the control group differed significantly from the low (p < 0.01) and high dose (p < 0.01) groups, but the low and high dose groups were not significantly different from each other (p > 0.2). Comparisons at individual times showed significant differences between control and high dose at 3+ months postexposure (all p-values <0.025) and marginally significant differences between low and high dose at 3+ months past exposure (all p-values between 0.015 and 0.075).
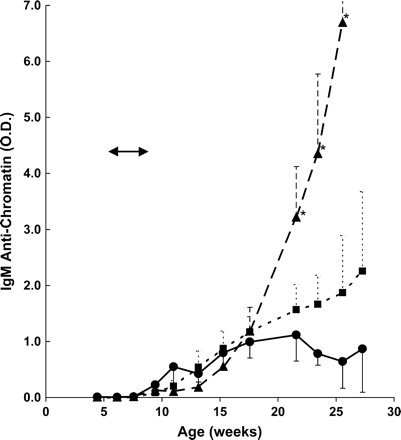
IgM anti-chromatin activity in MRL-lpr/lpr mice exposed to cigarette smoke. IgM serum autoantibodies were measured by ELISA. Cigarette smoke exposure period is marked by arrows and refers to doses of 0 (• —— •), 100 (▪- - - - -▪), or 250 (▴ – – – ▴) mg TPM/m3. Variances are SE, and asterisks indicate significant differences compared to control at individual time points shown.
We also examined IgM autoantibodies to denatured (d) DNA, a subset of which cross-react with the anti-phospholipid antibodies commonly seen in human SLE (Koike et al., 1982), as well as rheumatoid factor, IgM antibodies to IgG. The results of these assays are shown in Table 1. IgM anti-dDNA did not change between 4 and 8 weeks after the end of the smoke-exposure period, and this environmental exposure had no apparent effect on antibody levels. Rheumatoid factor in mice exposed to 250 mg smoke/m3 appeared substantially elevated at 8 weeks postexposure, although because of the large variation in the absolute values of these antibody activities among the mice, it did not reach statistical significance. Unlike the IgM anti-chromatin antibodies, rheumatoid factor did increase as the mice aged.
IgM Autoantibodies in MRL-lpr/lpr Mice after Exposure to Cigarette Smoke
IgM antibody activity (mean OD ± SD) | |||||||
|---|---|---|---|---|---|---|---|
| +8 weeksab | +16 weeksac | ||||||
| Smoke exposure (mg TPM/m3) | Anti-dDNA | Rheumatoid factor | Anti-dDNA | Rheumatoid factor | |||
| 0 | 4.13 ± 0.30 | 0.62 ± 0.64 | 4.12 ± 1.46 | 2.58 ± 3.48 | |||
| 100 | 4.04 ± 1.32 | 0.40 ± 0.46 | 4.33 ± 0.57 | 0.13 ± 0.13d | |||
| 250 | 3.06 ± 1.21 | 2.35 ± 3.48 | 7.07 ± 0.59 | 2.36 ± 2.48 | |||
IgM antibody activity (mean OD ± SD) | |||||||
|---|---|---|---|---|---|---|---|
| +8 weeksab | +16 weeksac | ||||||
| Smoke exposure (mg TPM/m3) | Anti-dDNA | Rheumatoid factor | Anti-dDNA | Rheumatoid factor | |||
| 0 | 4.13 ± 0.30 | 0.62 ± 0.64 | 4.12 ± 1.46 | 2.58 ± 3.48 | |||
| 100 | 4.04 ± 1.32 | 0.40 ± 0.46 | 4.33 ± 0.57 | 0.13 ± 0.13d | |||
| 250 | 3.06 ± 1.21 | 2.35 ± 3.48 | 7.07 ± 0.59 | 2.36 ± 2.48 | |||
Time after end of 4-week cigarette smoke exposure at the indicated dose, corresponding to 17.6 and 25.6 weeks of age.
n = 8/group.
n = 5–6/group.
The two mice with the highest rheumatoid factor activity at +8 weeks had died at +16 weeks.
IgM Autoantibodies in MRL-lpr/lpr Mice after Exposure to Cigarette Smoke
IgM antibody activity (mean OD ± SD) | |||||||
|---|---|---|---|---|---|---|---|
| +8 weeksab | +16 weeksac | ||||||
| Smoke exposure (mg TPM/m3) | Anti-dDNA | Rheumatoid factor | Anti-dDNA | Rheumatoid factor | |||
| 0 | 4.13 ± 0.30 | 0.62 ± 0.64 | 4.12 ± 1.46 | 2.58 ± 3.48 | |||
| 100 | 4.04 ± 1.32 | 0.40 ± 0.46 | 4.33 ± 0.57 | 0.13 ± 0.13d | |||
| 250 | 3.06 ± 1.21 | 2.35 ± 3.48 | 7.07 ± 0.59 | 2.36 ± 2.48 | |||
IgM antibody activity (mean OD ± SD) | |||||||
|---|---|---|---|---|---|---|---|
| +8 weeksab | +16 weeksac | ||||||
| Smoke exposure (mg TPM/m3) | Anti-dDNA | Rheumatoid factor | Anti-dDNA | Rheumatoid factor | |||
| 0 | 4.13 ± 0.30 | 0.62 ± 0.64 | 4.12 ± 1.46 | 2.58 ± 3.48 | |||
| 100 | 4.04 ± 1.32 | 0.40 ± 0.46 | 4.33 ± 0.57 | 0.13 ± 0.13d | |||
| 250 | 3.06 ± 1.21 | 2.35 ± 3.48 | 7.07 ± 0.59 | 2.36 ± 2.48 | |||
Time after end of 4-week cigarette smoke exposure at the indicated dose, corresponding to 17.6 and 25.6 weeks of age.
n = 8/group.
n = 5–6/group.
The two mice with the highest rheumatoid factor activity at +8 weeks had died at +16 weeks.
Proteinuria was determined 10 days after cessation of the smoke exposure protocol and at subsequent, approximately 2-week intervals. For several months after the smoke exposure period, an average of 50% of the control mice had detectable proteinuria at substantial levels (e.g., average ± SD of 113 ± 161 mg/dl at +4 weeks, n = 8) compared to background levels in normal mice of 0–10 mg/dl. With the treated mice, substantial proteinuria (≥100 mg/dl) during the first 4 weeks after exposure was detectable in only 20% and 0% of the mice exposed to 100 and 250 mg/m3 cigarette smoke, respectively. From then on, the percentage of mice with proteinuria increased in all three groups at about the same rate, so that at 12 weeks post-exposure the percentage of mice with proteinuria of at least 100 mg/dl was 75%, 60%, and 40% for the control group and the two smoke-exposed groups, respectively. However, by this time two mice in the control group and one mouse in the two smoke-exposed groups had died; after another month only half to one third of the mice were alive, all with proteinuria of 100–500 mg/dl. In order to maximize the duration of this study, no mice were sacrificed to directly evaluate kidney disease by histology, but the proteinuria and IgG autoantibody data are consistent with transient suppression of kidney disease due to cigarette smoke exposure.
In order to know whether the effects of cigarette smoke exposure on autoantibodies reflected a generalized effect on the humoral immune system, we determined the total IgG and IgM immunoglobulin levels in the mice. As shown in Fig. 3A, total IgG showed a large age-dependent increase, reaching a maximum of 23 mg/ml in 17.6-week-old mice, representing a >10-fold increase during the 4-month observation period followed by a substantial decline. IgG levels in smoke-exposed animals were indistinguishable from the controls in mice up to 6 months old, but continued to increase in mice previously exposed to 250 mg TPM/m3 from smoke, although this was not significantly different from the control group. IgM levels slowly increased throughout life, resulting in approximately a 10-fold increase from 4-week-old mice. However, there was no significant difference in IgM levels between the control and the smoke-exposed groups, although mice exposed to 100 mg TPM/m3 showed a substantial decline after 18 weeks of age (Fig. 3B). These results indicate that, in contrast to IgG and IgM autoantibodies, the overall humoral immune status of the mice was not affected in any significant way by exposure to cigarette smoke.
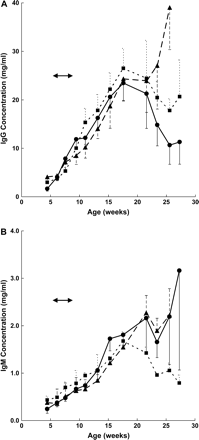
Immunoglobulin levels in MRL-lpr/lpr mice exposed to cigarette smoke. Total IgG (A) or IgM (B) immunoglobulins were measured by capture ELISA. Cigarette smoke exposure period is marked by arrows and refers to doses of 0 (• —— •), 100 (▪- - - - -▪), or 250 (▴ – – – ▴) mg TPM/m3. Variances are SE.
Association of Cigarette Smoking with Untreated SLE
In order to attempt to compare the mouse and human data without the added complication of the therapeutic interventions, we evaluated a subset of SLE patients with different smoking histories and in which immunosuppressive medications had not yet been introduced. This group of newly diagnosed patients could be considered to more closely match the smoking exposure of the mice used in the current study.
Of the 119 newly diagnosed patients, 88 were non-cigarette users at the time of diagnosis and 31 were cigarette smokers. We compared socio/economic characteristics, clinical symptoms, and laboratory findings at the time of diagnosis between these two patient groups. Cigarette users had smoked for an average of 12.3 ± 11.5 (± SD) years at an average of 0.8 ± 0.3 packs/day. As shown in Table 2, this group displayed significantly more pleuritis and peritonitis (p ≤ 0.03) compared to nonsmokers. Smokers also expressed more neuropsychiatric symptoms as measured by the neuro-SLEDAI and psychosis-8 indices as well as lupus headache (all p < 0.01). Nonsmokers tended to be more likely to present with evidence of nondeforming arthritis, but this did not reach significance. No other difference in symptomatic features between smokers and nonsmokers was detected. It should be noted that both the smokers and the nonsmokers displayed a remarkably high incidence of pleuritis; this symptom is often the reason the patients sought medical help before a diagnosis was made. Pleuritis generally responds to therapy, accounting for its significantly lower incidence in treated patients (Bombardier et al., 1992; Ghaussy et al., 2003).
Clinical Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a | p-value |
|---|---|---|---|
| pleuritisb | 74% | 51% | 0.034 |
| peritonitisb | 26% | 7% | 0.009 |
| arthritisb | 13% | 32% | 0.058 |
| psychosis-8c | 1.6 ± 3.2 | 0.3 ± 1.5 | 0.004 |
| neuro-SLEDAIc | 9.3 ± 8.3 | 5.3 ± 7.4 | 0.007 |
| lupus headache-8c | 4.9 ± 4.0 | 2.7 ± 3.8 | 0.008 |
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a | p-value |
|---|---|---|---|
| pleuritisb | 74% | 51% | 0.034 |
| peritonitisb | 26% | 7% | 0.009 |
| arthritisb | 13% | 32% | 0.058 |
| psychosis-8c | 1.6 ± 3.2 | 0.3 ± 1.5 | 0.004 |
| neuro-SLEDAIc | 9.3 ± 8.3 | 5.3 ± 7.4 | 0.007 |
| lupus headache-8c | 4.9 ± 4.0 | 2.7 ± 3.8 | 0.008 |
Includes nine former smokers.
Number is percent of patients displaying symptom; p-value calculated by Fisher's exact test.
Number is the average score ± SD; p-value calculated by Wilcoxon-Mann-Whitney test.
Clinical Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a | p-value |
|---|---|---|---|
| pleuritisb | 74% | 51% | 0.034 |
| peritonitisb | 26% | 7% | 0.009 |
| arthritisb | 13% | 32% | 0.058 |
| psychosis-8c | 1.6 ± 3.2 | 0.3 ± 1.5 | 0.004 |
| neuro-SLEDAIc | 9.3 ± 8.3 | 5.3 ± 7.4 | 0.007 |
| lupus headache-8c | 4.9 ± 4.0 | 2.7 ± 3.8 | 0.008 |
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a | p-value |
|---|---|---|---|
| pleuritisb | 74% | 51% | 0.034 |
| peritonitisb | 26% | 7% | 0.009 |
| arthritisb | 13% | 32% | 0.058 |
| psychosis-8c | 1.6 ± 3.2 | 0.3 ± 1.5 | 0.004 |
| neuro-SLEDAIc | 9.3 ± 8.3 | 5.3 ± 7.4 | 0.007 |
| lupus headache-8c | 4.9 ± 4.0 | 2.7 ± 3.8 | 0.008 |
Includes nine former smokers.
Number is percent of patients displaying symptom; p-value calculated by Fisher's exact test.
Number is the average score ± SD; p-value calculated by Wilcoxon-Mann-Whitney test.
Notable laboratory results are shown in Table 3. Smokers tended to have lower levels of IgG anti-DNA and anti-ribosomal P as well as less urinary protein but higher levels of IgM anti-phospholipid and rheumatoid factor antibodies. However, despite displaying three- to five-fold differences for several of these laboratory features, none reached statistical significance because of the large interpatient variance. No other differences in symptoms, signs, socioeconomic or demographic features between newly diagnosed smokers and nonsmokers were observed (Table 4).
Selected Laboratory Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31)a | Nonsmokers (n = 88)ab |
|---|---|---|
| anti-DNA | 29.4 ± 114.9 | 99.4 ± 211.6 |
| anti-ribosomal P | 9.3 ± 5.2 | 17.3 ± 35.8 |
| IgM Anti-phospholipid | 52.7 ± 182.1 | 9.1 ± 8.3 |
| IgM rheumatoid factor | 84.1 ± 214.2 | 29.7 ± 72.8 |
| urinary protein/creatine ratio | 0.5 ± 1.6 | 1.6 ± 3.4 |
Feature | Smokers (n = 31)a | Nonsmokers (n = 88)ab |
|---|---|---|
| anti-DNA | 29.4 ± 114.9 | 99.4 ± 211.6 |
| anti-ribosomal P | 9.3 ± 5.2 | 17.3 ± 35.8 |
| IgM Anti-phospholipid | 52.7 ± 182.1 | 9.1 ± 8.3 |
| IgM rheumatoid factor | 84.1 ± 214.2 | 29.7 ± 72.8 |
| urinary protein/creatine ratio | 0.5 ± 1.6 | 1.6 ± 3.4 |
Number is the average value ± SD. None of the comparisons were significantly different (p > 0.05) by Wilcoxon-Mann-Whitney test.
Includes nine former smokers.
Selected Laboratory Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31)a | Nonsmokers (n = 88)ab |
|---|---|---|
| anti-DNA | 29.4 ± 114.9 | 99.4 ± 211.6 |
| anti-ribosomal P | 9.3 ± 5.2 | 17.3 ± 35.8 |
| IgM Anti-phospholipid | 52.7 ± 182.1 | 9.1 ± 8.3 |
| IgM rheumatoid factor | 84.1 ± 214.2 | 29.7 ± 72.8 |
| urinary protein/creatine ratio | 0.5 ± 1.6 | 1.6 ± 3.4 |
Feature | Smokers (n = 31)a | Nonsmokers (n = 88)ab |
|---|---|---|
| anti-DNA | 29.4 ± 114.9 | 99.4 ± 211.6 |
| anti-ribosomal P | 9.3 ± 5.2 | 17.3 ± 35.8 |
| IgM Anti-phospholipid | 52.7 ± 182.1 | 9.1 ± 8.3 |
| IgM rheumatoid factor | 84.1 ± 214.2 | 29.7 ± 72.8 |
| urinary protein/creatine ratio | 0.5 ± 1.6 | 1.6 ± 3.4 |
Number is the average value ± SD. None of the comparisons were significantly different (p > 0.05) by Wilcoxon-Mann-Whitney test.
Includes nine former smokers.
Genetic and Socioeconomic Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a |
|---|---|---|
| Female | 100% | 100% |
| Ethnicity | 26% White, 71% Spanish, 3% Native American | 26% White, 65% Spanish, 5% Native American, 2% Asian, 2% Black |
| Family history of SLEb | 39% | 27% |
| Age of onsetc | 28.9 ± 18.0 | 30.6 ± 15.3 |
| Education (years)c | 11.1 ± 3.6 | 11.9 ± 1.8 |
| Employment (years)c | 9.2 ± 11.6 | 8.9 ± 9.6 |
| Family incomec | $27,000 ± $18,000 | $37,000 ± $44,000 |
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a |
|---|---|---|
| Female | 100% | 100% |
| Ethnicity | 26% White, 71% Spanish, 3% Native American | 26% White, 65% Spanish, 5% Native American, 2% Asian, 2% Black |
| Family history of SLEb | 39% | 27% |
| Age of onsetc | 28.9 ± 18.0 | 30.6 ± 15.3 |
| Education (years)c | 11.1 ± 3.6 | 11.9 ± 1.8 |
| Employment (years)c | 9.2 ± 11.6 | 8.9 ± 9.6 |
| Family incomec | $27,000 ± $18,000 | $37,000 ± $44,000 |
Note: There were no significant differences between smokers and nonsmokers in any of these parameters using the Wilcoxon-Mann-Whitney test or Fisher's exact test as appropriate.
Includes nine former smokers.
Self-reported.
Number is the average value ± SD.
Genetic and Socioeconomic Features of Newly Diagnosed SLE Patients
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a |
|---|---|---|
| Female | 100% | 100% |
| Ethnicity | 26% White, 71% Spanish, 3% Native American | 26% White, 65% Spanish, 5% Native American, 2% Asian, 2% Black |
| Family history of SLEb | 39% | 27% |
| Age of onsetc | 28.9 ± 18.0 | 30.6 ± 15.3 |
| Education (years)c | 11.1 ± 3.6 | 11.9 ± 1.8 |
| Employment (years)c | 9.2 ± 11.6 | 8.9 ± 9.6 |
| Family incomec | $27,000 ± $18,000 | $37,000 ± $44,000 |
Feature | Smokers (n = 31) | Nonsmokers (n = 88)a |
|---|---|---|
| Female | 100% | 100% |
| Ethnicity | 26% White, 71% Spanish, 3% Native American | 26% White, 65% Spanish, 5% Native American, 2% Asian, 2% Black |
| Family history of SLEb | 39% | 27% |
| Age of onsetc | 28.9 ± 18.0 | 30.6 ± 15.3 |
| Education (years)c | 11.1 ± 3.6 | 11.9 ± 1.8 |
| Employment (years)c | 9.2 ± 11.6 | 8.9 ± 9.6 |
| Family incomec | $27,000 ± $18,000 | $37,000 ± $44,000 |
Note: There were no significant differences between smokers and nonsmokers in any of these parameters using the Wilcoxon-Mann-Whitney test or Fisher's exact test as appropriate.
Includes nine former smokers.
Self-reported.
Number is the average value ± SD.
The nonsmokers included nine former smokers, people who stopped smoking 0–27 years (mean ± SD = 9 ± 11 years) prior to presenting to the clinic. Self-reported smoking history was a smoking habit of 0.7 ± 0.4 packs/day for 15.3 ± 15.9 (SD) years. This group was compared to never smokers and current smokers for clinical and laboratory features at the time of diagnosis. As shown in Table 5, eight of the nine former smokers (89%) had elevated IgG anti-DNA antibodies compared to 37% of never smokers and 29% of current smokers, a significant difference at the p < 0.01 based on a logistic regression model and adjusting or accounting for SLEDAI and SLICC scores, age, and age of onset. Former smokers tended to be older than never smokers at age of diagnosis (45 ± 26 years vs. 28 ± 26 years), although this difference did not reach statistical significance. The finding that former smokers were significantly more likely to have IgG anti-DNA antibodies than current smokers is remarkable and reminiscent of the murine lupus data (Fig. 1), which demonstrated that discontinuation of smoking was associated with the eventual exacerbation of humoral autoimmunity.
Anti-DNA Activity in Newly Diagnosed SLE Patients with Different Smoking Histories
Current smokers | Nonsmokers | Former smokers | |
|---|---|---|---|
| anti-DNA positive | 9 (29%) | 29 (37%) | 8 (89%)a |
| anti-DNA negative | 22 (71%) | 50 (63%) | 1 (11%)a |
Current smokers | Nonsmokers | Former smokers | |
|---|---|---|---|
| anti-DNA positive | 9 (29%) | 29 (37%) | 8 (89%)a |
| anti-DNA negative | 22 (71%) | 50 (63%) | 1 (11%)a |
Significantly different versus nonsmokers and current smokers at p < 0.01 by logistic regression.
Anti-DNA Activity in Newly Diagnosed SLE Patients with Different Smoking Histories
Current smokers | Nonsmokers | Former smokers | |
|---|---|---|---|
| anti-DNA positive | 9 (29%) | 29 (37%) | 8 (89%)a |
| anti-DNA negative | 22 (71%) | 50 (63%) | 1 (11%)a |
Current smokers | Nonsmokers | Former smokers | |
|---|---|---|---|
| anti-DNA positive | 9 (29%) | 29 (37%) | 8 (89%)a |
| anti-DNA negative | 22 (71%) | 50 (63%) | 1 (11%)a |
Significantly different versus nonsmokers and current smokers at p < 0.01 by logistic regression.
DISCUSSION
Insight into the capacity of cigarette smoking to affect signs and symptoms of SLE was obtained by determining the effect of tobacco smoke inhalation on humoral changes in autoimmune-prone mice and by analyzing untreated SLE patients with or without a smoking habit for features of autoimmunity. Conflicting previous results on the relationship between cigarette usage and SLE were recently subjected to a meta-analysis, indicating a small but significant association (Costenbader et al., 2004). However, the current study is the first in which SLE patients with different smoking histories were examined prior to therapeutic intervention, thereby minimizing possible confounding effects of smoking on medication effectiveness as discussed below. This is also the first study on the effects of cigarette smoke exposure on development of murine lupus, permitting a cross-species comparison with human disease. Contrary to our expectations, the results demonstrate that smoking suppressed IgG autoantibody development and that, after smoking was discontinued, autoantibody expression was augmented.
Development of autoantibodies in pre-autoimmune MRL-lpr/lpr mice showed a complex relationship to tobacco smoke exposure. Autoantibody appearance in control mice was first detectable at 8 weeks of age and continued to increase for the next 8 weeks, followed by a steady decline until death, presumably due to B-cell exhaustion and deposition of immune complexes associated with glomerulonephritis. Exposure of 6-week-old mice to cigarette smoke at 250 mg TPM/m3 air resulted in a 2-week delay in the appearance of IgG anti-chromatin antibodies, which remained significantly below controls 2 months after the end of the smoke-exposure period. However, unlike control mice, IgG anti-chromatin activity continued to increase in 4-month-old mice exposed to both the low and high dose smoke, reaching levels at 6 months significantly higher than the controls, 4 months after cessation of the smoke exposure protocol. It is unlikely that this difference in IgG anti-chromatin activity was due to differences in the amount of immune complex deposition because by this time all the mice displayed fulminant proteinuria. IgM anti-chromatin antibodies were unaffected during the first 3 months after the start of smoke exposure but also became significantly elevated over the control mice during the next few months. In contrast, nonimmune IgG and IgM as well as IgM anti-dDNA were not significantly altered by smoke exposure history. Overall, these results indicate that early-life exposure of lupus-prone mice to cigarette smoke suppresses IgG autoantibody development but not the age-dependent increase in total immunoglobulin. While this short-term cigarette smoke exposure had a long-lasting inhibitory effect on IgG autoantibody development, a rebound to higher levels occurred apparently after the suppressive effect of smoke inhalation had worn off.
SLE patients who smoked at the time of diagnosis were significantly more likely to have polyserositis and neuropsychiatric problems than nonsmokers, as well as more IgM but less IgG autoantibodies. These results are consistent with prior studies which found almost two-fold higher SLEDAI involving both neurological and nonneurological components in smokers than in nonsmokers (p < 0.001) (Ghaussy et al., 2003). Interestingly, almost all patients who discontinued smoking at an average of 9 years before diagnosis of SLE had elevated IgG anti-DNA antibodies compared to about one third of nonsmokers, a significantly different prevalence. These serological results are consistent with the murine data demonstrating suppressed IgG but not IgM anti-chromatin, anti-dDNA antibodies, and rheumatoid factor during and up to 8 weeks after smoke exposure, followed by elevated IgG autoantibodies 13–17 weeks after discontinuation of the smoke-exposure protocol. We suggest that patients with a smoking habit may have intrinsically worse disease than nonsmokers as previously concluded (Ghaussy et al., 2001), but smoking suppresses IgG autoantibodies; cessation of smoking results in elimination of this suppressive effect, resulting in emergence of IgG autoantibodies in both human and murine SLE.
Many compounds in cigarette smoke have been shown to alter immunity, and nicotine is implicated as the predominant immunosuppressive constituent (reviewed in Sopori, 2002). Rats continuously exposed in vivo to 330 μg nicotine/day for 3–4 weeks (equivalent to 30–40 cigarettes/day for humans) showed about a four-fold lower antibody response to a T-cell-dependent antigen, and an 80% reduction in the proliferative response of splenic T cells to anti-CD3 stimulation (Geng et al., 1995). This effect was associated with a blunted ability of nicotine-exposed T cells to mobilize calcium in response to receptor ligation (Geng et al., 1996) due to depletion of inositol 1,4,5-triphosphate-sensitive calcium stores by nicotine (Kalra et al., 2000). A smaller but still significant depression in the T-cell responsiveness was observed in rats that self-administered nicotine for 5 weeks, a condition more relevant to fluctuating human exposure levels of nicotine (Kalra et al., 2002).
While nicotine can be immunosuppressive, this effect wanes rapidly after discontinuation of this agent. The blunted immune response of rats exposed to nicotine for 4 weeks disappeared between 2 and 6 weeks after discontinuation of nicotine treatment (Geng et al., 1996). In the current study of autoimmune-prone mice, the effects of cigarette smoke exposure endured for 2 months, well beyond the reported capacity of nicotine to suppress components of the peripheral immune system. While it is possible that other components of cigarette smoke with immunomodulatory properties such as acrolein, benzo[a]pyrene, or benzene (Sopori, 2002) may be involved, they are present in mainstream smoke at one tenth to one hundredth the concentration of nicotine (Surgeon General Report, U.S. Department of Health and Human Services, Washington, DC, 1989). Considering the high concentration and well-known T-cell inhibitory capacity of nicotine, we propose that it may cause prolonged immunosuppression by acting in the thymus, reducing the output and/or function of autoreactive T cells.
Unlike other mouse models of SLE, autoimmunity expression in MRL-lpr/lpr mice is highly sensitive to a functional thymus after birth (Theofilopoulos and Dixon, 1985). Neonatal thymectomy results in greatly diminished anti-DNA antibodies, proteinuria, lymphadenopathy, and glomulonephritis as manifested 5–6 months later (Kalra et al., 2004; Steinberg et al., 1980), and it is likely that autoreactive T cells are continually emigrating from the thymus throughout life in this strain. Recent studies demonstrated that nicotine inhibits T-cell maturation in the thymus (Middlebrook et al., 2002), apparently by interacting with nicotinic acetylcholine receptors (Kuo et al., 2002; Talib et al., 1993), blocking T-cell development at the CD4+CD8+ stage (Middlebrook et al., 2002). Autoreactive T-cell output may have been suppressed in smoke-exposed mice by this mechanism, thereby reducing autoantibody production by autoreactive B cells, which are dependent on T-cell help for autoantibody expression (Gilkeson et al., 1992; Sobel et al., 1994). The rebound to supra-elevated levels by 3–4 months may be due to the response of a T helper cell-starved population of autoreactive B cells to resumption of autoreactive T-cell output from the thymus. Consistent with this scenario was the observation that IgM autoantibodies, which in young MRL-lpr/lpr mice are derived from T-cell-independent, polyclonal activation of intrinsically defective B cells (Merino et al., 1993), were not affected by smoke exposure. The late-life enhancement in the IgM, together with the IgG-anti-chromatin response in the smoke-exposed mice, may be related to the report that IgM autoantibodies in MRL-lpr/lpr mice can be driven by CD4+ T-cell help (Merino et al., 1993). The finding that total, nonimmune immunoglobulin was not significantly affected by smoke exposure suggests that the intrinsic defect(s) in MRL-lpr/lpr mice leading to autoreactive T-cell production (Hahn, 2002; Theofilopoulos and Dixon, 1987) may be particularly sensitive to the inhibitory effects of nicotine.
Human studies also suggest a generalized suppressive state of the immune system as a result of cigarette smoking. Normal subjects who smoked cigarettes for at least 1 year had 25% lower serum IgG levels than nonsmokers (p < 0.001) but similar IgM levels (Ferson et al., 1979). CD4+ T cells were significantly lower in heavy smokers (Miller et al., 1982) and in smokers from a black population between 20 and 50 years old, which normalized 2–5 years after discontinuing smoking (Tollerud et al., 1991). Smoking may suppress certain inflammatory diseases as well. Since first reported (Harries et al., 1982), numerous studies have demonstrated a negative correlation between smoking and ulcerative colitis, with the risk of ulcerative colitis in smokers being one-third (Jick and Walker, 1983) to one-quarter (Benoni and Nilsson, 1987; Logan et al., 1984) that of nonsmokers at the time of diagnosis. In one study of 154 patients, onset of ulcerative colitis occurred in 70% of the patients within an average of 5.7 years after discontinuation of smoking (Motley et al., 1987). Ulcerative colitis is considered an inflammatory disease typically benefited by treatment with immunosuppressive agents, implying a likewise protective effect of smoking. Administration of nicotine for 6 weeks by transdermal patch produced significant symptomatic improvement of ulcerative colitis (Pullan et al., 1994); transdermal nicotine for 4 weeks also suppressed provoked cutaneous inflammation (Mills et al., 1997). These and other studies (reviewed in Holt, 1987) indicate that long-term smoking is associated with suppression of the adaptive, T-cell-dependent immune system.
The conclusion that nicotine intake due to cigarette smoking suppresses the immune system may seem at odds with studies demonstrating a positive correlation between smoking and immune-mediated diseases. Smoking has been reported to be more prevalent in patients with rheumatoid arthritis (odds ratio (OR) = 1.31 (Karlson et al., 1999), OR = 13.5 in heavy smokers (Hutchinson et al., 2001), OR = 2.0 in elderly women (Criswell et al., 2002)), in patients with Graves' disease (OR = 1.9 (Prummel and Wiersinga, 1993)), especially Europeans (OR = 2.4 (Tellez et al., 1992)), in patients with primary biliary cirrhosis (OR = 2.0 (Parikh-Patel et al., 2001) or 2.4 (Howel et al., 2000)), and in patients with SLE (OR = 0.9–6.7, average = 1.5 (Costenbader et al., 2004)). Therefore, even in the context of a generalized immunosuppressive effect of nicotine, some autoimmune diseases may be aggravated by smoking due to other components of cigarette smoke besides nicotine.
However, the interpretation of these studies is unclear. Most patients with chronic rheumatologic disease are undergoing immunosuppressive therapy, and smoking may alter pharmacokinetics by increasing plasma clearance, suppressing gastrointestinal absorption, or potentiating hepatic drug metabolism (Miller, 1990). Polycyclic aromatic hydrocarbons, which are major components of cigarette smoke, can lower the effective therapeutic levels of numerous medications by induction of hepatic mixed-function oxidases, particularly the CYP1A2 isoenzyme, which can metabolize many drugs to inactive forms (Schein, 1995). As a result, the effective concentration of the medication is lowered. For example, smokers required twice the dosage of theophylline compared to nonsmokers to achieve similar therapeutic benefit or a three- to four-fold dose reduction during smoking abstinence to avoid adverse affects (Holt, 1987; Miller et al., 1982). Reduction in effective therapeutic efficacy for smokers was observed with tacrin, flecainide, propoxyphene, propranolol, diazepam, chlordiazepoxide, and pentazocine (Schein, 1995), and induction of liver enzymes in smokers has been shown to increase the metabolism of imipramine, meprobamate, estrogens, phenylbutazone, theophylline, and warfarin (Miller, 1989). In a retrospective cohort study of cutaneous lupus patients treated with chloroquine or hydroxychloroquine, skin disease resolved in 53% of nonsmokers compared to 18% of smokers, consistent with the view that smoking reduces effective therapeutic levels of antimalarials in SLE patients (Rahman et al., 1998). This would suggest that the association between cigarette smoking and disease activity in SLE may be the result of smokers being undertreated with anti-inflammatory or immunosuppressive medications due to smoking-induced reduction in effective drug levels, and that SLE patients who smoke may require higher therapeutic doses.
In the current study we sought to minimize the role of smoking-induced suppression of effective drug concentration by retrospectively evaluating a subset of patients prior to initiating treatment. The lower levels of IgG but not IgM autoantibodies in smokers and the apparent return to elevated anti-DNA in former smokers is consistent with the observations in murine lupus that smoking suppressed T-cell-dependent autoimmunity in these patients. However, SLE patients who smoked tended to have worse symptomatic disease, as previously reported (Ghaussy et al., 2003), consistent with the view that any immunosuppressive effect of smoking is insufficient to influence the clinical course of established SLE, as previously concluded (Benoni et al., 1990). It is possible that the high prevalence of smoking in SLE patients reflects a craving of patients with incipient and/or intrinsically worse disease to self-treat, consistent with the finding that the prevalence of cigarette smoking in SLE patients does not significantly change after diagnosis and physician counseling (Freemer and Criswell, 2003). A subclinical benefit may be obtained from the immunosuppressive or anti-inflammatory effects of smoking.
The authors certify that all research involving human subjects was done under full compliance with all government policies and the Helsinki Declaration.
This project was supported in part by Tobacco Settlement funds of the University of New Mexico School of Medicine, NIEHS Center grant P30-012072 (SWB), NIH ES06334 (RLR), and NIH NS35708 (WLS). The excellent assistance of Vanessa N. Arteaga and Lee F. Blair is also appreciated. Informed consent of each patient was obtained, and this study was approved by the institutional review board. Conflict of interest: none declared.
References
Aarden, L. A., deGroot, E. R., and Feltkamp, T. E. W. (
Bell, S. A., Hobbs, M. V., and Rubin, R. L. (
Benoni, C., and Nilsson, A. (
Benoni, C., Nilsson, A., and Nived, O. (
Bombardier, C., Gladman, D. D., Urowitz, M. B., Caron, D., and Chang, C. H. (
Burlingame, R. W., and Rubin, R. L. (
Burlingame, R. W., and Rubin, R. L. (
Collins, R. J., Neil, J. C., and Wilson, R. J. (
Costenbader, K. H., Kim, D. J., Peerzada, J., Lockman, S., Nobles-Knight, D., Petri, M., and Karlson, E. W. (
Criswell, L. A., Merlino, L. A., Cerhan, J. R., Mikuls, T. R., Mudano, A. S., Burma, M., Folsom, A. R., and Saag, K. G. (
Feltkamp, T. E., Kirkwood, T. B., Maini, R. N., and Aarden, L. A. (
Ferson, M., Edwards, A., Lind, A., Milton, G. W., and Hersey, P. (
Finch, G. L., Lundgren, D. L., Barr, E. B., Chen, B. T., Griffith, W. C., Hobbs, C. H., Hoover, M. D., Nikula, K. J., and Mauderly, J. L. (
Freemer, M. M., and Criswell, L. A. (
Geng, Y., Savage, S. M., Johnson, L. J., Seagrave, J., and Sopori, M. L. (
Geng, Y., Savage, S. M., Razani-Boroujerdi, S., and Sopori, M. L. (
Ghaussy, N. O., Sibbitt, W., Jr., Bankhurst, A. D., and Qualls, C. R. (
Ghaussy, N. O., Sibbitt, W. L., Jr., and Qualls, C. R. (
Gilkeson, G. S., Spurney, R., Coffman, T. M., Kurlander, R., Ruiz, P., and Pisetsky, D. S. (
Gladman, D., Ginzler, E., Goldsmith, C., Fortin, P., Liang, M., Urowitz, M., Bacon, P., Bombardieri, S., Hanly, J., Hay, E., et al. (
Hahn, B. H. (
Harries, A. D., Baird, A., and Rhodes, J. (
Hawker, G., Gabriel, S., Bombardier, C., Goldsmith, C., Caron, D., and Gladman, D. (
Hochberg, M. C. (
Howel, D., Fischbacher, C. M., Bhopal, R. S., Gray, J., Metcalf, J. V., and James, O. F. (
Hutchinson, D., Shepstone, L., Moots, R., Lear, J. T., and Lynch, M. P. (
Jick, H., and Walker, A. M. (
Kalra, R., Singh, S. P., Kracko, D., Matta, S. G., Sharp, B. M., and Sopori, M. L. (
Kalra, R., Singh, S. P., Pena-Philippides, J. C., Langley, R. J., Razani-Boroujerdi, S., and Sopori, M. L. (
Kalra, R., Singh, S. P., Savage, S. M., Finch, G. L., and Sopori, M. L. (
Karlson, E. W., Lee, I. M., Cook, N. R., Manson, J. E., Buring, J. E., and Hennekens, C. H. (
Koike, T., Tomiaka, H., and Kumagi, A. (
Kuo, Y., Lucero, L., Michaels, J., DeLuca, D., and Lukas, R. J. (
Logan, R. F., Edmond, M., Somerville, K. W., and Langman, M. J. (
Mauderly, J. L., Gigliotti, A. P., Barr, E. B., Bechtold, W. E., Belinsky, S. A., Hahn, F. F., Hobbs, C. A., March, T. H., Seilkop, S. K., and Finch, G. L. (
Merino, R., Iwamoto, M., Fossati, L., and Izui, S. (
Merkel, P. A., Chang, Y., Pierangeli, S. S., Harris, E. N., and Polisson, R. P. (
Middlebrook, A. J., Martina, C., Chang, Y., Lukas, R. J., and DeLuca, D. (
Miller, L. G. (
Miller, L. G. (
Miller, L. G., Goldstein, G., Murphy, M., and Ginns, L. C. (
Mills, C. M., Hill, S. A., and Marks, R. (
Motley, R. J., Rhodes, J., Ford, G. A., Wilkinson, S. P., Chesner, I. M., Asquith, P., Hellier, M. D., and Mayberry, J. F. (
Parikh-Patel, A., Gold, E. B., Worman, H., Krivy, K. E., and Gershwin, M. E. (
Peacock, J. L., Cook, D. G., Carey, I. M., Jarvis, M. J., Bryant, A. E., Anderson, H. R., and Bland, J. M. (
Prummel, M. F., and Wiersinga, W. M. (
Pullan, R. D., Rhodes, J., Ganesh, S., Mani, V., Morris, J. S., Williams, G. T., Newcombe, R. G., Russell, M. A., Feyerabend, C., Thomas, G. A., et al. (
Rahman, P., Gladman, D. D., and Urowitz, M. B. (
Reidenberg, M. M., Drayer, D. E., Lorenzo, B., Strom, B. L., West, S. L., Snyder, E. S., Freundlich, B., and Stolley, P. D. (
Rubin, R. L., Balderas, R. S., Tan, E. M., Dixon, F. J., and Theofilopoulos, A. N. (
Schein, J. R. (
Selgrade, M. K., Cooper, G. S., Germolec, D. R., and Heindel, J. J. (
Sibbitt, W. L., Jr., Schmidt, P. J., Hart, B. L., and Brooks, W. M. (
Sobel, E. S., Kakkanaiah, V. N., Kakkanaiah, M., Cheek, R. L., Cohen, P. L., and Eisenberg, R. A. (
Steinberg, A. D., Roths, J. B., Murphy, E. D., Steinberg, R. T., and Raveche, E. S. (
Talib, S., Okarma, T. B., and Lebkowski, J. S. (
Tan, E. M., Cohen, A. S., Fries, J. F., Masi, A. T., McShane, D. J., Rothfield, N. F., Schaller, J. G., Talal, N., and Winchester, R. J. (
Tellez, M., Cooper, J., and Edmonds, C. (
Theofilopoulos, A. N., and Dixon, F. J. (
Theofilopoulos, A. N., and Dixon, F. J. (
Tollerud, D. J., Brown, L. M., Blattner, W. A., Mann, D. L., Pankiw-Trost, L., and Hoover, R. N. (
Author notes
*Department of Molecular Genetics and Microbiology, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131; †Department of Mathematics and Statistics, University of New Mexico, Albuquerque, New Mexico 87131; ‡Lovelace Respiratory Research Institute, Albuquerque, New Mexico 87108; §College of Pharmacy Toxicology Program, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131; and ¶Departments of Internal Medicine and Neurology, University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131

{kind=link}
{kind=link}
{kind=link}
Comments