Article Text

Abstract
Objective Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterised by heterogeneous clinical manifestations, autoantibody production and epigenetic dysregulation in T cells. We sought to investigate the epigenetic contribution to the development of cutaneous manifestations in SLE.
Methods We performed genome-wide DNA methylation analyses in patients with SLE stratified by a history of malar rash, discoid rash or neither cutaneous manifestation, and age, sex and ethnicity matched healthy controls. We characterised differentially methylated regions (DMRs) in naïve CD4+ T cells unique to each disease subset, and assessed functional relationships between DMRs using bioinformatic approaches.
Results We identified 36 and 37 unique DMRs that contribute to the epigenetic susceptibility to malar rash and discoid rash, respectively. These DMRs were primarily localised to genes mediating cell proliferation and apoptosis. Hypomethylation of MIR886 and TRIM69, and hypermethylation of RNF39 were specific to patients with SLE with a history of malar rash. Hypomethylation of the cytoskeleton-related gene RHOJ was specific to patients with SLE with a history of discoid rash. In addition, discoid rash-specific hypomethylated DMRs were found in genes involved in antigen-processing and presentation such as TAP1 and PSMB8. Network analyses showed that DMRs in patients with SLE with but not without a history of cutaneous manifestations are associated with TAP-dependent processing and major histocompatibility-class I antigen cross-presentation (p=3.66×10−18 in malar rash, and 3.67×10−13 in discoid rash).
Conclusions We characterised DNA methylation changes in naïve CD4+ T cells specific to malar rash and discoid rash in patients with SLE. These data suggest unique epigenetic susceptibility loci that predispose to or are associated with the development of cutaneous manifestations in SLE.
- Systemic Lupus Erythematosus
- Gene Polymorphism
- Autoimmune Diseases
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
We identified epigenetic susceptibility loci for cutaneous involvement in SLE using DNA methylation profiles in naïve CD4+ T cells.
Differentially methylated regions localized to genes mediating cell proliferation and apoptosis contribute to the epigenetic susceptibility to cutaneous involvement in SLE.
Cutaneous involvement in SLE is characterized by differential DNA methylation in genes involved in TAP-dependent processing and MHC-I antigen cross-presentation.
Novel targets that can help to better understand cutaneous manifestations in SLE have been identified.
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterised by autoantibody production and heterogeneous clinical manifestations. The aetiology of SLE remains incompletely understood, however there is increasing evidence for a role of DNA methylation changes in the pathogenesis of SLE.1 DNA methylation is a lineage-specific epigenetic mechanism with an integral role in the immune system. This DNA modification, which typically refers to the methylation of the 5′ cytosine carbon of cytosine-guanine (CG) dinucleotides, is often a transcriptionally repressive mark able to alter gene accessibility and chromatin structure.2 Through this effect, DNA methylation is capable of mediating cell differentiation and immune function.3 Indeed, differentiation of naïve CD4+ T cells to TH1, TH2 and TH17 effector subsets is imparted by demethylation of IFNγ, IL-4/IL-5/IL-13 and IL-17A/IL-17F genes, respectively.3 Importantly, aberrancies in DNA methylation can cause significant dysregulation of the immune system and have been associated with several autoimmune conditions including SLE.1 ,2 ,4–18 We previously reported evidence linking type I interferon hyper-responsiveness in patients with SLE to transcriptional poising induced by DNA hypomethylation in naïve CD4+ T cells.1
In this study, we explore DNA methylation changes in naïve CD4+ T cells in patients with SLE with a history of malar rash or discoid rash to identify patterns of epigenetic susceptibility that are specific to patients with a history of these cutaneous manifestations. We identified differentially methylated (DM) sites unique to either cutaneous manifestation in SLE. In addition, we associate manifestation-specific differentially methylated regions (DMRs) to pathways related to environmental stress response, apoptosis and proliferation, and antigen processing and presentation.
Materials and methods
Patient and control demographic information
This study included three independent groups of patients with SLE and healthy matched controls. Each group consisted of eight patients with SLE with a history of malar rash, discoid rash or neither, and eight age (±5 years), sex and ethnicity matched healthy controls (see online supplementary table S1). All patients fulfilled the American College of Rheumatology classification criteria for SLE.19 The American College of Rheumatology classification criteria for SLE met in each patient, and disease activity scores and criteria measured using the SLE Disease Activity Index and background medications at the time of enrolment in this study are shown in online supplementary tables S2 and S3. Patients and healthy controls included in this study signed an informed consent, and were recruited from the University of Michigan, Oklahoma Medical Research Foundation and the Henry Ford Health System.
Naïve CD4+ T cell DNA extraction
From each study participant, 80 mL of whole blood was collected then subjected to Ficoll-gradient centrifugation (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) to isolate peripheral blood mononuclear cells. Naïve CD4+ T cells were then isolated from peripheral blood mononuclear cells by negative selection magnetic bead cell separation (indirect labelling) using the Naïve CD4+ T Cell Isolation kit II (Miltenyi Biotec, Cambridge, Massachusetts, USA). The purity of the isolated naïve CD4+ T cells was confirmed >95% using fluorochrome-conjugated antibodies targeting CD4 and CD45RA. DNA was then extracted using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, California, USA) and bisulfite converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, California, USA) for subsequent DNA methylation analysis.
DNA methylation studies
Genome-wide DNA methylation analysis was performed using the Infinium HumanMethylation450K BeadChip array (Illumina, San Diego, California, USA). DNA methylation levels were assessed at 485 577 methylation sites throughout the human genome, across 96% of UCSC cytosine-phosphate-guanine island (CpG islands) and 99% of RefSeq genes with an average of 17 sites per gene covering enhancers, promoter regions, 5′untranslated region (UTRs), 3′UTRs and gene bodies.
Statistical and bioinformatic analyses
Genome-wide DNA methylation analyses were performed using GenomeStudio methylation module V.1.9.0 (Illumina) as described previously.1 Probe signal intensities were derived from raw image intensities then normalised using non-CG probes. Background subtraction was then performed based on unhybridised negative-control probe intensities. The normalised, background-subtracted signal intensities were used to calculate β values that represent DNA methylation levels on a scale of 0 to 1. Differential DNA methylation was calculated between patients with SLE and their respective matched controls within the malar rash, discoid rash or neither cutaneous involvement group using the GenomeStudio Illumina custom model described previously.1 Probes with a single nucleotide polymorphism (SNP) within 10 bp of the 3′ probe end and probes with a detection p value ≥0.05 were excluded from the analysis. Differentially methylated sites were then filtered to include CG sites with a methylation difference (|Δβ|) ≥0.10 between patients and controls, and a differential methylation score (|DiffScore|) > 22, which corresponds to a p value ≤0.01 after correction for multiple testing using a Benjamini and Hochberg false discovery rate of 5%. Hypomethylated regions (hypo-DMR) and hypermethylated regions (hyper-DMR) were identified as clusters of at least two respective hypomethylation or hypermethylation sites <500 bp of each other using the clusterMaker function and Bump Hunter package for the R statistical programming language.20 Regions were then filtered to exclude all hypo-DMR or hyper-DMR overlapping between different cutaneous manifestation groups to identify DMRs unique to each group. Network analyses were performed using GeneMANIA software with networks of gene-gene interactions based on attributes, coexpression, colocalisation, genetic interactions, pathways, physical interactions, predicted interactions and shared protein domains.21 ,22 Network analysis results are presented as false discovery rate corrected p values.
Results
Global DNA methylation patterns in cutaneous SLE
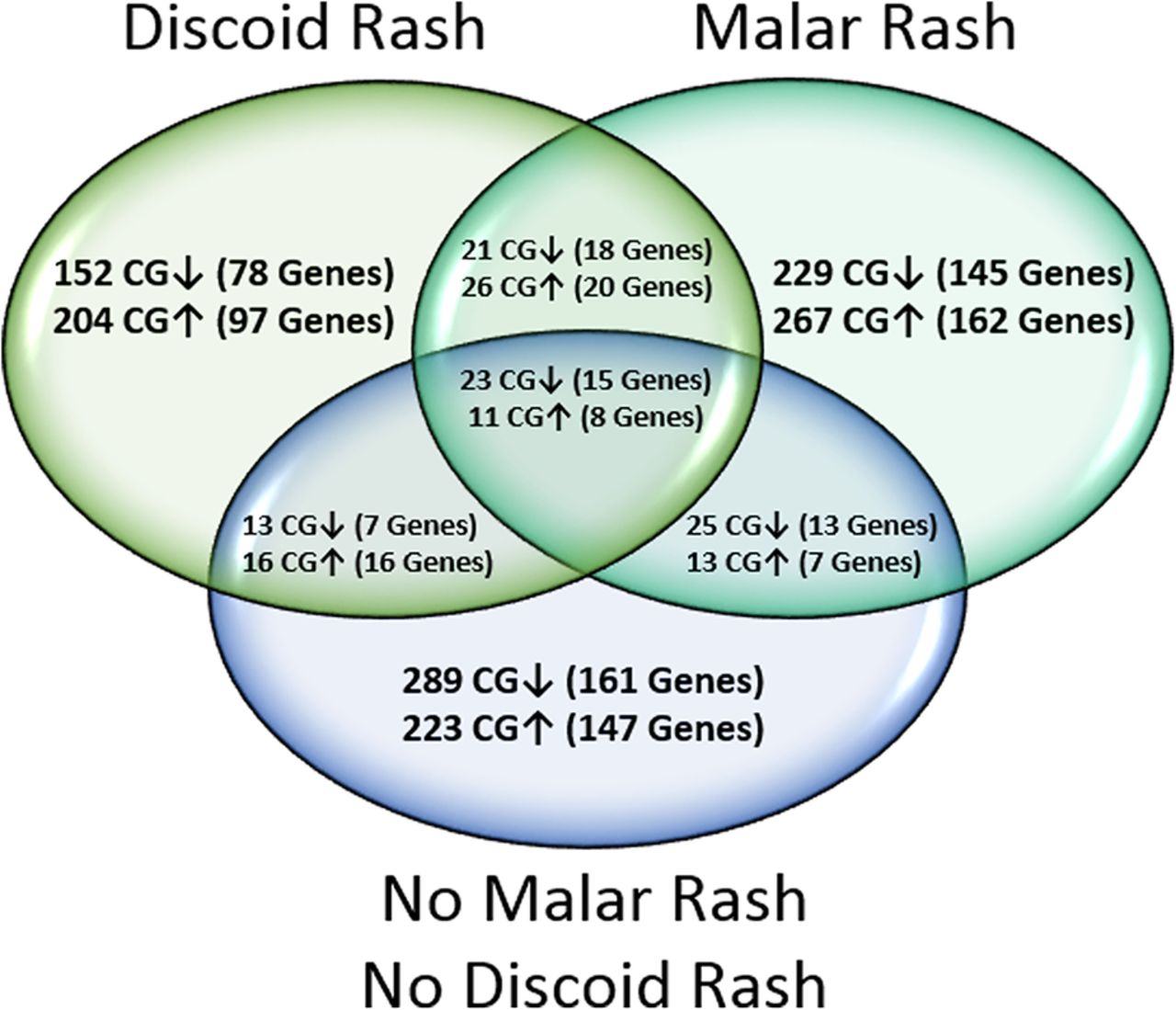
Genome-wide DNA methylation profiles of peripheral blood naïve CD4+ T cells were created for SLE with a history of malar rash, discoid rash and neither cutaneous involvement by comparing DNA methylation levels at 485 577 sites between patients and matched healthy controls. We identified 615 DM sites in the malar group, 466 DM sites in the discoid group and 613 DM sites in the SLE group with neither cutaneous involvement. The majority of DM sites identified in each group were manifestation-specific, and present only in one group but not the remaining two groups (figure 1). Common to all groups, however, was a consistent hypomethylation in interferon-regulated genes (see online supplementary table S4). DNA methylation changes unique to malar rash, discoid rash or the SLE group with neither cutaneous involvement were subjected to further analysis to identify DNA methylation changes that predispose to specific cutaneous manifestations in SLE.

Venn diagram depicting the distribution of differentially methylated (DM) CG sites (|Δβ|≥0.10) and associated genes between patients with systemic lupus erythematosus with a history of malar rash, discoid rash or neither cutaneous involvement, compared with healthy matched controls. Hypermethylated and hypomethylated CG sites are shown with ↑ and ↓ arrows, respectively.
The greatest methylation differences unique to malar rash were found in GNAS (Δβ=−0.27) encoding the G-protein α-stimulatory subunit and an intergenic locus at 7p22.2 (Δβ=0.33) (table 1, online supplementary table S4). In patients with discoid rash, the greatest unique methylation differences were found in KNDC1 (Δβ=−0.21) which has a potential role in p53-mediated cell cycle arrest, an intergenic locus at 3q29 (Δβ=0.26), and HLA-DRB6 (Δβ=0.26) (table 1, online supplementary table S4).23 The greatest unique methylation differences in patients with SLE with no cutaneous involvement are found in NFYA (Δβ=−0.28) encoding a subunit of the NF-Y transcription factor and in EXOC7 (Δβ=0.33) which encodes a component of the exocyst complex which is involved in exocytosis and membrane remodelling (table 1, online supplementary table S4).
Differential methylation analysis results showing the 10 most hypomethylated and hypermethylated CG sites (|Δβ|≥0.10) specific to (A) malar rash, (B) discoid rash or (C) neither cutaneous involvement in patients with systemic lupus erythematosus
SLE manifestation-specific DMR analysis
Next, we identified genomic regions with extensive DNA methylation differences in patients with SLE with malar rash, discoid rash or no cutaneous involvement compared with their respective controls. For each SLE group studied, unique DMRs were defined as genomic ranges of at least two CG sites within a 500 bp window that are uniquely hypomethylated or hypermethylated in each patient group subset compared with healthy controls, but not in the two remaining SLE groups. Our data for malar rash SLE showed 14 hypomethylated regions (hypo-DMRs) and 22 hypermethylated regions (hyper-DMRs) (table 2). The most extensive region contains 13 hypermethylated sites (mean Δβ=0.15) in LY6G5C, a leucocyte antigen-6 gene of the major histocompatibility (MHC) class III genomic region. In patients with discoid rash, we show 17 hypo-DMRs and 20 hyper-DMRs with the most extensive DMR containing a cluster of 11 hypermethylated sites (mean Δβ=0.16) within RNF39 (HZF), a gene of the MHC class I genomic region encoding haematopoietic zinc finger protein (table 2).
Unique differentially methylated regions (DMRs) in naïve CD4+ T cells from patients with systemic lupus erythematosus with a history of (A) malar rash, (B) discoid rash or (C) neither cutaneous involvement
Common to both cutaneous rashes are DMRs in an intergenic region in 7p22.3 and in PRIC285 which encodes a transcriptional coactivator of peroxisome proliferator-activated receptor (PPAR)α and PPARγ (see online supplementary table S5).24 ,25 In patients with SLE with no cutaneous involvement, we identified 28 hypo-DMRs and 16 hyper-DMRs with the most extensive region containing 15 hypomethylated sites (mean Δβ=−0.15) within TNXB, a gene of the MHC class III genomic region encoding tenascin XB extracellular matrix glycoprotein (table 2). In all three patient subset groups, we identified a shared hypo-DMR in IFI44L (malar rash mean Δβ=−0.20, discoid rash mean Δβ=−0.16 and neither cutaneous involvement mean Δβ=−0.15) and GSTT1 (malar rash mean Δβ=−0.14, discoid rash mean Δβ=−0.15 and neither cutaneous involvement mean Δβ=−0.15) (see online supplementary table S5). In addition, we identified a shared hyper-DMR in GSTTP1 (malar rash mean Δβ=0.16, discoid rash mean Δβ=0.15 and neither cutaneous involvement mean Δβ=0.16).
Gene network analysis of manifestation-specific DMRs
We then performed gene network analyses to assess relationships between the manifestation-specific DMR genes. Results for malar rash and discoid rash hypo-DMR analyses showed enrichment in genes functioning in antigen peptide transporter (TAP)-dependent antigen processing and presentation of exogenous peptides via MHC class I (p=3.66×10−18 in malar and p=3.67×10−13 in discoid) (figure 2, table 3, online supplementary table S6). This pathway was not significantly enriched in genes with unique hypo-DMR in patients with neither cutaneous involvement (p>0.05) (figure 2, online supplementary table S6). Network analyses were also performed for hyper-DMR genes, yet no functions were enriched in either discoid rash or neither cutaneous involvement groups. However, malar hyper-DMR genes were highly enriched in functions associated with type I interferon response (p=1.11×10−21) (see online supplementary figure S1, online supplementary table S7). It should be noted, however, that these hypermethylated interferon-related DMR genes are more specifically enriched in negative regulation of type I interferon production (p=4.83×10−4) and are different from the interferon-regulated genes that constitute the interferon signature known to be hypomethylated in SLE. Indeed, SLE interferon-signature genes were consistently hypomethylated in all three SLE groups in this study (see online supplementary table S4).
Network analysis results are shown for genes with unique hypomethylated regions (hypo-DMRs) in naïve CD4+ T cells from patients with systemic lupus erythematosus (SLE) with a history of malar rash, discoid rash or neither cutaneous involvement

{kind=link}
{kind=link}
Network analysis results showing relationships among genes with hypomethylated regions (hypo-DMRs) specific to patients with systemic lupus erythematosus with a history of (A) malar rash, (B) discoid rash or (C) neither cutaneous involvement. Manifestation-specific hypo-DMR genes and network-associated genes are represented by blue and grey nodes, respectively. The lines connecting the nodes depict gene-gene relationships based on coexpression (purple), colocalisation (blue), genetic interactions (green), physical interactions (pink), and shared protein domains (yellow). Line thickness is relative to the strength of the gene-gene relationship. All network analyses were performed using GeneMANIA software. DMR, differentially methylated region.
Discussion
DNA methylation is a stable and heritable epigenetic modification that has a considerable effect on naïve CD4+ T cell differentiation and immune function. Aberrancies in DNA methylation are found in several autoimmune diseases including SLE.15 ,26 Furthermore, robust demethylation of interferon-regulated genes in naïve CD4+ T cells in SLE has been associated with a hyper-responsive type I interferon signature in patients with SLE.1 Not surprisingly, demethylation of these same interferon-regulated genes was common to all SLE groups with malar, discoid or neither cutaneous manifestation (see online supplementary table S4). In this study, we further investigate the important link between naïve CD4+ T cells and SLE pathogenesis in a clinical manifestation-specific manner with an in-depth analysis of aberrant DNA methylation in naïve CD4+ T cells from patients with SLE with a history of malar rash or discoid rash. This is accomplished with a case-control study design involving independent genome-wide DNA methylation analyses for SLE with a history of malar rash, discoid rash or neither cutaneous involvement. In each analysis, patients with SLE in each of the manifestation groups were compared with healthy controls carefully matched for age, sex and ethnicity on a one-by-one basis. We then excluded DM sites that were common between the manifestation groups to characterise DNA methylation changes that are unique to only malar rash or discoid rash.
Our global DNA methylation profiles showed distinguishable manifestation-specific differences between each SLE group with 80.7% unique DM sites (229 hypomethylated and 267 hypermethylated CGs) for malar rash, and 76.4% unique DM sites (152 hypomethylated and 204 hypermethylated CGs) for discoid rash. In addition, we identified 14 hypo-DMRs and 22 hyper-DMRs unique to malar rash and 17 hypo-DMRs and 20 hyper-DMRs unique to discoid rash. Interestingly, several DMRs in either cutaneous manifestation are associated with genes mediating cell proliferation and apoptosis pathways. In patients with a history of malar rash, the most extensive malar rash DMR consists of six hypomethylated CG sites (mean Δβ=−0.12) in the promoter region of precursor microRNA MIR886 (VTRNA2-1).27 Hypomethylation of the VTRNA2-1 promoter has been shown to downregulate the interferon-inducible phosphorylated RNA-dependent protein kinase which in turn regulates eIF2α and Nuclear Factor kappa B (NF-κB) signalling pathways pivotal to determining cell survival or apoptosis.28–30 We also found a hypo-DMR in TRIM69 (two hypomethylated CG sites, mean Δβ=−0.10) whose overexpression has been shown to induce apoptosis in murine models. In patients with a history of discoid rash, we report a DMR in the DNA damage response genes RHOJ (2 hypomethylated CG sites, mean Δβ=−0.17) and HZF (11 hypermethylated CG sites, mean Δβ=0.16) which are also involved in cell survival/apoptosis pathway commitment. Indeed, the haematopoietic zinc finger encoded by HZF is a key cofactor of p53 tumour suppressor and serves as a critical switch promoting cell-cycle arrest over the alternative apoptosis pathway.31 ,32 Taken together, both cutaneous manifestations are associated with unique DMRs in genes that influence environmental stress response pathways and cell fate decisions. It is unclear what role these DMRs have in cutaneous manifestations in SLE. However, they may provide insight into the impaired apoptotic cell clearance found in the epidermis of patients with cutaneous lupus erythematosus and in germinal centres of patients with SLE.33 ,34
Our DMR analyses also revealed a relationship between SLE cutaneous manifestations and antigen processing and presentation. In particular, SLE with discoid rash is associated with hypo-DMR in PSMB8 (two hypomethylated sites, mean Δβ=−0.13), encoding a subunit of the immunoproteasome involved in processing peptides for MHC-I loading. A hypo-DMR was also found in TAP1 (four hypomethylated sites, mean Δβ=−0.11) which encodes a key subunit of the transporter associated with antigen processing complex, TAP. In monocytes and dendritic cells, the TAP complex transports phagosome-processed exogenous peptides to the endoplasmic reticulum to bind and present on MHC-I in a process known as cross-presentation.35 Interestingly, our network analyses of unique hypo-DMR in each cutaneous patient subset group showed enrichment in genes functioning in the TAP-dependent antigen processing and presentation of exogenous peptide antigen via MHC-I (malar rash p=3.66×10−18, discoid rash p=3.67×10−13). MHC-I cross-presentation is commonly used by antigen-presenting cells, and it is unclear what role it may have in CD4+ T cells in SLE. One possibility is that these changes represent epigenetic susceptibility loci that can manifest in other cell types that might be more directly involved in antigen cross presentation. The other possibility is that cross-presentation genes and pathways are aberrantly active in CD4+ T cells in a subset of patients with SLE and might play a different role in the pathogenesis of SLE that is independent of their classical known role in antigen cross-presentation. It is also worth noting that T cells have been previously shown to present antigens on MHC class I and MHC class II,36 and whether this is involved in the pathogenesis of cutaneous SLE remains to be further examined.
In this study, we performed an extensive investigation of naïve CD4+ T cell DNA methylation changes associated with malar rash and discoid rash in SLE. In either of the cutaneous manifestations, we characterised aberrant DNA methylation profiles and identified DMRs in multiple genes that impact environmental stress response and cell fate. In addition, hypo-DMRs of malar rash and discoid rash are enriched in genes involved in the pathway for TAP-dependent exogenous antigen processing and MHC-I cross-presentation. Importantly, we revealed several novel targets of aberrant DNA methylation in naïve CD4+ T cells that might aid in our understanding of SLE and its cutaneous manifestations. Future studies and replication efforts with a larger sample size to detect smaller DNA methylation changes between patients and controls, and to examine other cell subsets relevant to SLE might uncover additional epigenetic susceptibility loci for cutaneous SLE manifestations.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors All authors listed contributed and fulfil authorship criteria as below: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI097134.
Competing interests None declared.
Ethics approval Institutional review boards at our institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.