Article Text

Abstract
Background SLE is likely triggered by gene–environment interactions. We have shown that most SLE-associated haplotypes encompass genomic regions enriched for epigenetic marks associated with enhancer function in lymphocytes, suggesting genetic risk is exerted through altered gene regulation. Data remain scarce on how epigenetic variance contributes to disease risk in paediatric SLE (pSLE). We aim to identify differences in epigenetically regulated chromatin architecture in treatment-naive patients with pSLE compared with healthy children.
Methods Using the assay for transposase-accessible chromatin with sequencing (ATACseq), we surveyed open chromatin in 10 treatment-naive patients with pSLE, with at least moderate disease severity, and 5 healthy children. We investigated whether regions of open chromatin unique to patients with pSLE demonstrate enrichment for specific transcriptional regulators, using standard computational approaches to identify unique peaks and a false discovery rate of <0.05. Further analyses for histone modification enrichment and variant calling were performed using bioinformatics packages in R and Linux.
Results We identified 30 139 differentially accessible regions (DAR) unique to pSLE B cells; 64.3% are more accessible in pSLE than healthy children. Many DAR are found in distal, intergenic regions and enriched for enhancer histone marks (p=0.027). B cells from adult patients with SLE contain more regions of inaccessible chromatin than those in pSLE. In pSLE B cells, 65.2% of the DAR are located within or near known SLE haplotypes. Further analysis revealed enrichment of transcription factor binding motifs within these DAR that may regulate genes involved in pro-inflammatory responses and cellular adhesion.
Conclusions We demonstrate an epigenetically distinct profile in pSLE B cells when compared with healthy children and adults with lupus, indicating that pSLE B cells are predisposed for disease onset/development. Increased chromatin accessibility in non-coding genomic regions controlling activation of inflammation suggest that transcriptional dysregulation by regulatory elements controlling B cell activation plays an important role in pSLE pathogenesis.
- lupus erythematosus, systemic
- autoimmune diseases
- B-lymphocytes
Data availability statement
Data are available in a public, open access repository. ATACseq data sets generated and analysed for this manuscript are uploaded into Sequence Read Archive, per protocol, accession number PRJNA884097 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA884097). Adult ATACseq data set is publicly available at Sequence Read Archive, accession number PRJNA290920 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA290920).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Lupus is an autoimmune disease that can affect major organs of the body; it is well-known that there are differences in gene expression between children with lupus and those without. However, genetic variations and the environment can also alter the immune response and result in disease.
WHAT THIS STUDY ADDS
Epigenomic sequencing techniques used in our study revealed differences in transcription regulation, and increased accessibility, of genes important in activation of the immune system and the proinflammatory response in children with lupus compared with adults with lupus and children without lupus.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study provides a more comprehensive picture of what may make children vulnerable to the development of lupus, as well as the differences in response to therapies between patients, and allows us to design new treatments that may be safer and more effective in children than current therapies.
Introduction
SLE is a complex disease thought to be triggered by gene–environment interactions. It is often marked by periods of disease flare and remission, and optimal treatment remains challenging. Pediatric-onset SLE (pSLE) accounts for up to 20% of all patients with SLE and can have a more severe and debilitating course than adult SLE; paediatric patients experience a shorter time to first intensive care admissions and are typically treated with multiple immunosuppressive medications to achieve disease control.1 2 We have shown that much of the SLE genetic risk identified in several genome-wide association studies (GWAS) lies in functional, non-coding3 regions of the genome that are responsible for the regulation of transcription.
Functional elements that regulate and coordinate transcription are typically found in regions of ‘open’ chromatin4 5 and are marked by specific epigenetic signatures consisting of specific histone marks.6 7 These histone marks are abundant in non-coding regions of the genome, affect other functional elements and have promoter or repressor properties, leading to altered chromatin accessibility and ultimately affecting gene expression. Coit et al8 9 have shown that gene expression and regulation are specific to distinct cell types and that cell-specific expression is a feature of chronic diseases such as SLE. Dozmorov et al10 demonstrated that adult SLE B cells express different genes than a population of monocytes from the same patients. It has been well-established that B cells are of central importance in disease pathogenesis in SLE and contribute toward disease flares (reviewed in Rose and Dörner11). These data also suggest that SLE may develop secondary to aberrant transcriptional and epigenetic control in B cells, resulting in the cells’ inability to coordinate and regulate transcription across the genome.
One way to investigate epigenetic changes in any chronic disease is to examine chromatin architecture, including accessible regions, using assays such as DNAse I hypersensitivity assays or assays for transposase-accessible chromatin with sequencing (ATACseq). Pioneered by Buenrostro et al,12 ATACseq allows for the surveying of the epigenome for differences in chromatin architecture and accessibility. Traditional methods such as DNAse I hypersensitivity assays typically require a large number of cells (often over 1×108) to achieve adequate sequencing depth. Initially, ATACseq required as few as 50 000 cells; the advent of newer sequencing technologies now allow ATACseq at the single-cell level (single-cell ATACseq),13 making ATACseq a more feasible assay to generate high-quality data for pSLE research.
To date, only one other group has used ATACseq to investigate epigenetic influences on disease development in adult SLE B cells: Scharer et al14 described disordered chromatin architecture in adult SLE B cells when compared with healthy adult B cells. In particular, they noted increased chromatin accessibility for transcription factors important in B cell activation and differentiation in SLE B cells, in contrast to increased accessibility to transcription factors important in processes associated with regulation of transcription in healthy adult B cells. Gensterblum et al15 used ATACseq to investigate chromatin accessibility in a newly defined T cell subset thought to be important in predicting disease activity in adult SLE. Data remain scarce on how epigenetic variance contributes to disease risk in pSLE or how epigenetic features of the genome are influenced by genetic variants. We aim to identify differences in epigenetically regulated chromatin architecture in treatment-naive patients with pSLE compared with healthy children.
Methods
Patient enrolment
We obtained approval to conduct this prospective, observational study from the Northwell Health Institutional Review Board (IRB# HS16-0017) and informed consent from all enrolled patients and/or parents. Assent was obtained from children between the ages of 7 and 17 years in accordance with IRB policy. All patients seen at the Pediatric Rheumatology Clinic at Cohen Children’s Medical Center between February 2016 and April 2018 with paediatric-onset lupus (diagnosed prior to their 19th birthdays), who had not received prior treatment for lupus, were eligible for enrolment into this prospective, observational study. All enrolled patients fulfilled the 1997 American College of Rheumatology (ACR) classification criteria16 and had a disease activity index score (SLEDAI) of over 4 at diagnosis, indicating at least moderate disease severity.17 Healthy children were identified and enrolled from our General Pediatric and Adolescent Medicine clinics. Children were excluded if they had another autoimmune or chronic inflammatory disease, were obese (body mass index above 30), were acutely ill with fever or on antibiotics for a recent infection, or on corticosteroids for any reason.
B cell isolation and enrichment
At time of enrolment, 20 mL of whole blood was collected from each patient (treatment-naive patients with lupus and healthy children) and processed within 4 hours of arrival in our laboratory. B cells were obtained by negative selection using the STEMCELL Technologies (Vancouver, BC, Canada) B cell enrichment kit (product #19054), using anti-CD3 (BioLegend #300469), anti-CD14 (BioLegend #301853), anti-CD16 (BioLegend #302059) and positive selection using anti-CD19 (BioLegend #302212) antibodies. Cells obtained in this manner were 95% CD19+ when assessed by flow cytometry.
Assays for transposase-accessible chromatin with sequencing (ATACseq) preparation and statistical analysis
After B cell enrichment, 50 000 cells were transferred into sterile DNAse/RNAse free Eppendorf tubes on ice and washed three times with cold phosphate-buffered saline (PBS, ThermoFisher Scientific, Massachusetts, USA). Resulting pellets were then kept at −80°C until ready for shipment to the Jarvis Laboratory for ATACseq library preparation.
ATACseq consists of three major steps: preparation of nuclei, insertion of Tn5 transposases and DNA purification, and PCR amplification across sequencing adapters. We used the ATACseq protocol described by Buenrostro et al.12 Briefly, cells were thawed and lysed using cold lysis buffer (10 nM Tris-Cl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IPEGAL CA-630, ThermoFisher Scientific). Immediately post-lysis, nuclei were spun at 4°C at 500 g for 10 min and re-suspended in the transposase reaction mix (25 μL 2× TD buffer, 2.5 μL transposase (Illumina, San Diego, USA) and 22.5 μL nuclease-free water). The transposition reaction was carried out at 37°C for 30 min; immediately after, the Qiagen Minelute kit was used to purify the sample (Qiagen, Maryland, USA). Post-purification, library fragments were amplified using 1× NEBnext PCR mastermix and 1.25 uM PCR primer1 and barcoded PCR primer2 (New England Biolabs, Massachusetts, USA). The following thermocycler conditions were used: 5 min at 72°C, 30 s at 98°C, cycling for 10 s at 98°C, 30 s at 63°C and 1 min at 72°C. We monitored the PCR using quantitative PCR to reduce genome copy (GC) and size bias in the reactions by amplifying the full libraries for five cycles, then adding 5 μL of the PCR to 10 μL PCR cocktail with SyBR green (ThermoFisher Scientific) at a final concentration of 0.6×. To determine the additional number of cycles needed for the remaining 45 μL reaction, we ran the 15 μL reaction for 20 cycles and stopped amplification prior to saturation. We then plotted linear Rn vs cycle to determine the cycle number corresponding to 1/3 of the maximum fluorescent intensity. AMPure XP beads (Beckman Coulter Life Sciences, California, USA) were used to purify the libraries, yielding a final library of 17.5 μL. Libraries were then sent to the Genomics and Bioinformatics Core facility at the University at Buffalo for 50 bp paired-end sequencing on the Illumina HiSeq 2500 platform.
Data analysis
Conventional computational approaches were used for peak calling. In brief, raw data (FASTQ) files had adapters trimmed using Trim Galore18 and quality control using FASTQC.19 Files were then aligned to the human genome (hg19) using BBMap, a splice-aware aligner for DNA and RNA sequencing reads, with mapping quality (MAPQ)>30, and sorted using Samtools.20 21 Only uniquely mapped and non-redundant reads were used for downstream analysis. Peak calling was performed using Model-Based Analysis for chromatin immunoprecipitation with sequencing (ChIP-seq, MACS2) using its default settings.22 Differential accessibility analysis was performed using DiffBind23 and centred around peak summits, extending 1000 base pairs in each direction. Peaks with false discovery rate <0.05 were considered significantly differentially accessible between pSLE and healthy children. Histone modification analysis was performed using the Roadmap/ENCODE database and gene ontology (GO) analysis for biological function with the GO database. Motif detection was performed using PScan-ChIP and HOMER.24 25 Peaks were annotated using ChIPpeakAnno and ChIPseeker. Nucleosome positioning analysis was performed using NucleoATAC in Python and NucHunter, a Java-based application. GO enrichment analysis was performed using the publicly available Gene Ontology enRIchment analysis and visualisation tool (GORILLA),26 and further reduced using the Reduce Visualise Gene Ontology tool (REVIGO) based on semantic similarity measures.27
Comparison of ATACseq data from adult and paediatric B cells
We compared ATACseq data from adult SLE and pSLE B cells. In brief, we downloaded and queried ATACseq data from CD19+ B cells of 15 adult patients with lupus and 51 healthy adults from the publicly available NCBI Sequence Read Archive (SRA, dataset GSE118256, 14). No further annotation of these patients was available. Quality inspection of the downloaded FASTQ files was performed using FASTQC.19 The reports for all FASTQ files were combined (by condition: SLE and healthy) using MULTIQC.28 No red flags requiring remediation were found. Alignment to the human GRCh38 reference genome was performed using Bowtie2 with its default parameters.29 Major single nucleotide polymorphisms (SNPs) from 1K genomes were included with the reference genome.30 Post-alignment, SAM files were converted to BAM files, sorted and indexed using Samtools.21 The resulting BAM files were shifted using deepTools2 (left-most fragments were shifted 4 bases to the right and right-most shifted 5 bases to the left).31
Similar methodology was used to call peaks and compare DARs between adult and pSLE B cells as was used to identify DARs between paediatric patients and healthy children. DARs from the adult patients with SLE were subtracted from pSLE DARs, using Bedtools, to focus on the differences attributable to disease pathology.32 Motif analysis was again performed using PScan-ChIP and HOMER24 25 and annotated with ChIPseeker.
Patient and public involvement
Plans for dissemination of the results of this study will involve presentation at conferences with patients and families in attendance, in addition to speaking with patients and support groups. We also plan to work with patients and families to discuss further dissemination plans beyond the scientific community and acknowledge their contributions where appropriate.
Results
Patient demographics
Ten newly diagnosed, treatment-naive patients with pSLE and five healthy children were enrolled in this study (table 1). There were seven female (70%) and three male (30%) patients in the pSLE cohort, ranging from 7 to 17 years of age. There were four healthy females (80%) and one healthy male (20%), ranging from 9 to 18 years of age. In the pSLE cohort, the mean age of disease onset was 12.5 (range 7–17) years of age, and mean SLEDAI 13 (range 6–24). All patients had a SLEDAI over 4, indicating at least moderate disease activity at time of enrolment. Lupus manifestations of rash and arthritis were present in 90% of the patients, and biopsy-proven lupus nephritis in 50%. All patients had serologies positive for ANA and anti-double-stranded DNA (dsDNA) autoantibodies, which are specific for a diagnosis of lupus. All patients also had low complement levels, which are a marker for active disease (table 1).
Patient demographics
B cells from paediatric patients with lupus exhibit a unique chromatin architecture
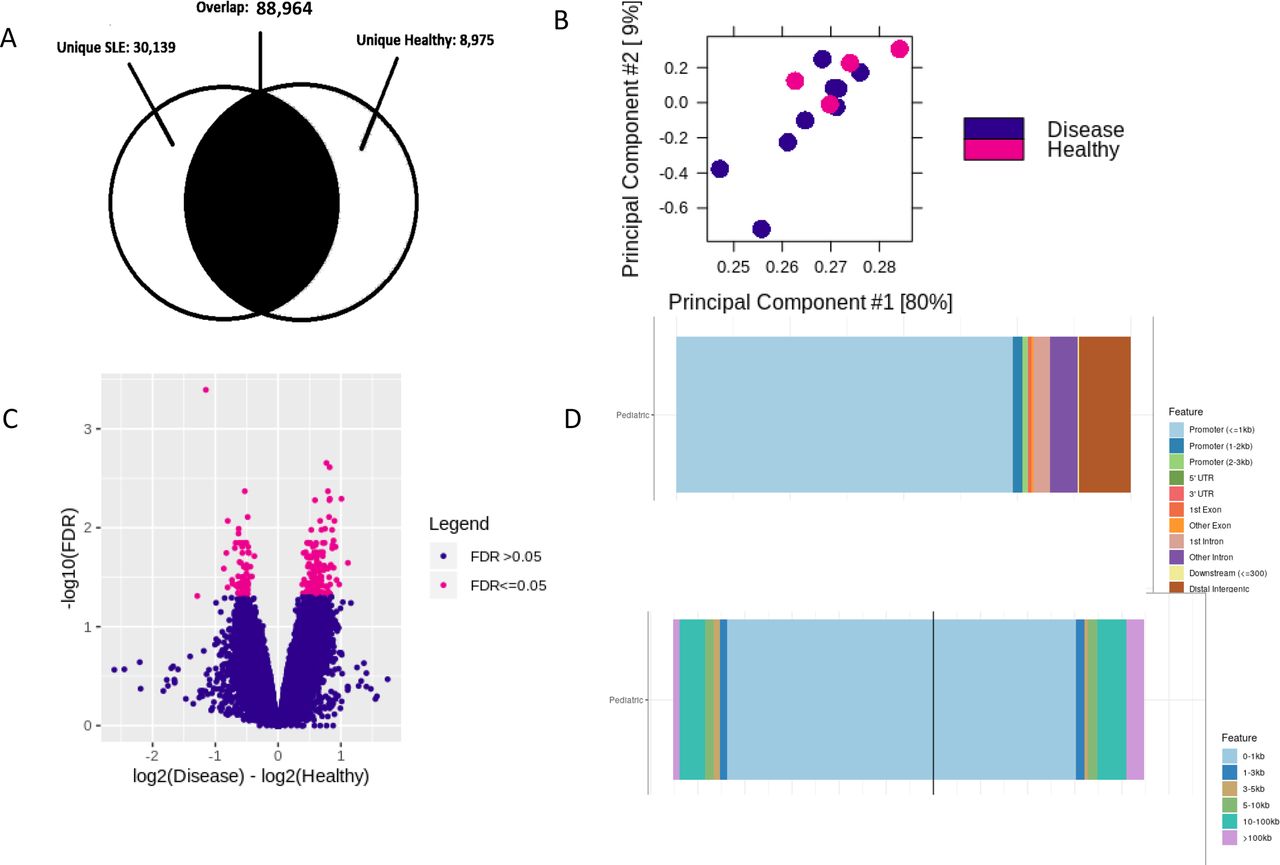
Differential peak analysis identified 30 139 uniquely accessible sites in patients with pSLE (figure 1) when compared with the B cells of healthy control children. Of these sites, 64.3% are more accessible in patients with pSLE than healthy children, but there were also regions that demonstrated decreased accessibility in the B cells from patients with pSLE when compared with those from healthy children (figure 1C).

Unique chromatin architecture in paediatric-onset lupus B cells compared with B cells from healthy children. Peaks distinct from B cells from healthy children tend to be located distally from transcription start sites. (A) Venn diagram showing unique peaks in paediatric patients with lupus (30 139) and healthy children (8975). (B) Principal component analysis plot showing distribution of paediatric lupus (purple points) and healthy children (pink points) samples. Note that the lupus samples in closer proximity to healthy children samples have milder disease severity. (C) Differentially accessible regions in paediatric lupus B cells compared with healthy children (false discovery rate (FDR)<0.05). Pink plotted points to the right of 0 on the x-axis indicate increased chromatin accessibility in paediatric patients with lupus. Pink plotted points to the left of 0 on the x-axis indicate potentially less accessible chromatin in paediatric patients with lupus. (D) Top histogram showing that over 75% of unique peaks in paediatric patients with lupus are located 10–100 kb or more from transcription start sites. Bottom histogram showing that the peaks that are located around transcription start sites are enriched for transcription factor binding loci.
There were 50.34% of the differentially accessible regions (DAR) in pSLE found in distal, intergenic regions of the genome; only 7.7% of the DAR are located directly in promoters or exons, mostly belonging to genes that regulate cell homeostasis or cell cycle activities. A select few appear to be involved in the proinflammatory response (eg, interferon-related genes) or cellular adhesion (eg, chemokines). Further analyses of these open regions using ChipEnrich revealed that 65.4% of these unique peaks are located between 10 and 100 kb and above from the nearest transcription start site (figure 1D), implying that many transcription factors may be acting within distal enhancers to regulate transcription.
Areas of increased chromatin accessibility in B cells in paediatric lupus are associated with dysregulated transcription of the proinflammatory response
We previously identified 46 SNPs from GWAS in adults with SLE that lie in distal intergenic regions and may regulate transcription.3 We compiled a list of another 325 SNPs from more recent GWAS in adult SLE that are enriched for enhancer function and thus may regulate transcription.33–35 Using NucleoATAC to interrogate broad peaks obtained from patients with pSLE, and Bedtools intersect to identify nucleosome positions that overlap with DAR, generated 158 nucleosome positions that lie within 10–100 kb of SNPs used to tag SLE risk loci identified by GWAS and our previous study.3 33–35 We then used single nucleotide polymorphisms annotator (SNiPA)36 to determine the haplotypes defined by these SNPs (r2<0.9). The majority of SNPs investigated that may contribute to lupus disease risk and their associated linkage disequilibrium blocks (eg, SLE haplotype) are present within 65.2% of the DARs from our pSLE data, implying perturbation of the affected genomic region and altered transcription factor binding affinity. Local or cis expression quantitative trait loci (eQTLs) were found in 43.2% of the SLE haplotypes containing DAR from patients with pSLE; similarly, 34.3% of these haplotypes contained trans eQTLs.
When the DAR that are more accessible in pSLE B cells were interrogated in the Roadmap/ENCODE database, we found that these regions were enriched for enhancer histone marks H3K4me2, H3K4me3 and H3K27ac (p=0.027) when compared with randomly generated regions of the genome. This finding is consistent with our previous analysis of SLE genetic risk loci compiled from several GWAS, which are also enriched for the same histone marks.3 Subsequent analysis with ChipEnrich found these DAR to contain consensus binding sites for 42 transcription factors that may be accessible in patients with pSLE but not healthy children, including 6 that have been identified in previous GWAS as conferring disease risk: nuclear receptor subfamily 4 group A member 1 (Nr4a1), growth factor independent 1b transcriptional repressor (GFI1b), eomesodermin (Eomes), signal transducer and activator of transcription 3 (STAT3), ETS proto-oncogene 1 (Ets1) and ETS like-1 protein (Elk1).37–40 ChipEnrich also identified polycomb repressive complex 2 subunit (SUZ12) and enhancer of zeste-homolog 2 (EZH2) to be enriched within the DAR. SUZ12, enhancer of zeste-homolog 1 (EZH1) and EZH2 have been shown to regulate recruitment of the transcriptional machinery to transcription start sites of genes important to the activation of the proinflammatory response.41 Indeed, using Pscan-ChIP, a web server that finds over-represented transcription factor binding motifs described by motif profiles available in databases such as JASPAR or TRANSFAC, and their correlated sequences in ChIP-seq experiments,24 several DAR in pSLE B cells displayed enrichment for binding motifs for transcription factors, including STAT3, nuclear factor kB (NFkB) and peroxisome proliferator activated receptor gamma (PPARg). No difference in accessibility was observed for a control motif, not found to be enriched in either pSLE or healthy B cells. Thus, our data identified activation of the proinflammatory response in pSLE B cells manifested in changes in chromatin architecture surrounding specific transcription factor binding motifs.
Gene ontology enrichment analysis of 3864 genes expressed within DAR of pSLE B cells, compared against a background of all protein-coding genes (from BioMart Ensembl), was performed using GORILLA. This revealed 129 different biologic processes present within the DAR, most notably cellular activation in the immune response, regulation of cell proliferation and cellular responses to external stimuli (eg, interferon).
Areas of increased chromatin accessibility in B cells from paediatric patients with lupus are associated with SLE haplotypes
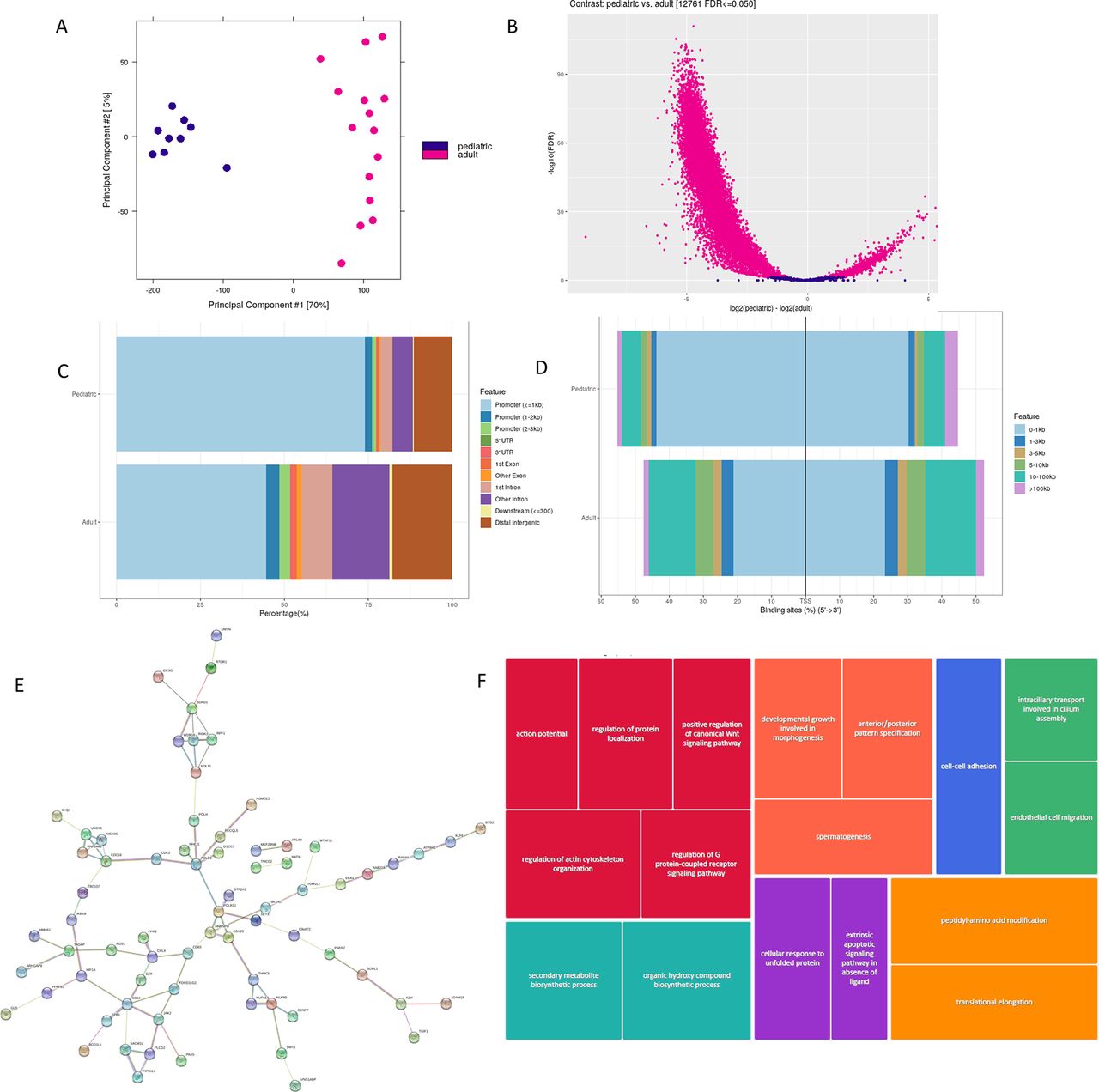
As mentioned above, we downloaded ATACseq data from B cells of adult patients with lupus and healthy adults in the GSE118235 dataset, publicly available on NCBI SRA,14 and applied the pipeline described above for analysis of ATACseq data in our paediatric samples, comparing the results between the two cohorts. Differential peak analysis revealed 12 761 DARs, with more regions of chromatin inaccessibility in B cells from adult SLE than pSLE (figure 2). We also found more peaks located over 100 kb from the nearest transcription start site in adult SLE than in pSLE. Peaks located in closer proximity to transcription start sites had less enrichment for transcription factor binding loci in adult lupus B cells than paediatric lupus B cells.

Unique chromatin architecture in paediatric-onset lupus B cells compared with adult lupus B cells. (A) Principal component analysis plot showing distribution of paediatric lupus (darker points) and adult lupus (lighter points) samples. (B) Differentially accessible regions in paediatric lupus B cells compared with adult lupus B cells (false discovery rate (FDR)<0.05). Lighter plotted points to the right of 0 on the x-axis indicate increased chromatin accessibility in paediatric patients with lupus. Lighter plotted points to the left of 0 on the x-axis indicate potentially less accessible chromatin in paediatric patients with lupus. Note that there appears to be more closed chromatin than open chromatin in adult patients with lupus compared with paediatric lupus. (C) Histogram showing that there are fewer peaks located distally and intergenic in paediatric lupus B cells than adult lupus B cells. (D) Histogram showing that peaks found near transcription start sites in paediatric lupus B cells have more enrichment of transcription factor binding loci than adult lupus. (E) Protein–protein interaction network of genes interacting with enhancers that overlap differentially accessible binding sites between paediatric and adult lupus B cells (plotted in String). (F) REVIGO gene ontology tree map highlighting upregulated pathways in paediatric lupus but not adult lupus B cells. Shades of grey demonstrate superclusters, and box sizes indicate the strength of the p value (larger size reflects higher statistical significance).
Moreover, there were 65.2% of the DAR in patients with pSLE located within known SLE haplotypes, compared with 39.7% of DAR from B cells of adult SLE patients (p=0.02). Gene ontology enrichment analysis of 1661 genes expressed within adult SLE DARs within known SLE haplotypes revealed 130 different biologic processes, most notably cellular activation in the immune response, and complement activation. The same analysis of overlapping DARs between adult SLE and pSLE within known SLE haplotypes revealed 209 genes belonging to cellular processes important in immune cell activation, including JAK/STAT and toll-like receptor (TLR) signalling, and cellular adhesion (figure 2E,F).
Genes involved in the pro-inflammatory response and cellular adhesion are more accessible in B cells in paediatric lupus than in adult lupus B cells
Using the Integrated Genome Viewer,42 a high-performance visualisation tool for interactive exploration of large genomic datasets, many loci with increased accessibility in patients with pSLE are located near start sites of their adjacent genes (figure 3). Fifty-five per cent of pSLE DAR located within SLE haplotypes lie within 1 kbp of genes involved in cell proliferation, activation of the adaptive immune response, activation of the innate immune response and protein degradation, eg, interferon regulatory factor-5 (IRF5) (figure 3B), STAT4 and tumour necrosis factor alpha-induced protein 3 (TNFAIP3). Polymorphisms in the above genes have been shown to be associated with disease pathogenesis in adult SLE.43 44 STAT4 and IRF5 are important regulators of type I interferons; peripheral B cells in patients with SLE display a prominent, highly reproducible type I interferon gene signature. Similar changes were not observed in the accessibility of IRF5 or STAT4 in B cells from healthy children.

{kind=link}
{kind=link}
{kind=link}
Loci with increased chromatin accessibility in paediatric patients with lupus can cluster around transcription start sites of genes important in the proinflammatory response, which may then be poised for activation. (A) Fragment size distribution as obtained with NucleoATAC. (B) Broad peaks from paediatric patients with lupus within DAR in SLE haplotype containing IRF5 are clustered near the transcription start site. This haplotype is also enriched for enhancer marks, indicating that the likelihood of transcription of IRF5 is very high in paediatric lupus B cells. (C) Representative figure of three-dimensional chromatin conformation around STAT4. The triangle represents a topographically associated domain, and the thin rectangle is the location of the SNP used to tag the risk locus. Note that the thin rectangle appears to overlap the topographically associated domain, indicating close contact with the chromatin loops and suggesting that STAT4 is poised for activation in paediatric lupus B cells. DAR, differentially accessible regions; IRF5, interferon regulatory factor-5; NFR, nucleosome-free region; STAT4, signal transducer and activator of transcription 4.
Further interrogation of the DAR residing within SLE haplotypes revealed 57 genes, including those important in cell-to-cell adhesion (cadherins) and chemokine-mediated signalling, eg, C-C motif chemokine ligand 4 (CCL4), which have been suggested to play a role in SLE pathogenesis in adults.45 46 Using Juicebox,47 an integrated viewer of Hi-C data, we discovered that several of these genes, including those involved in JAK/STAT signalling, were in close three-dimensional contact with chromatin loops in B cell lymphoma cell lines, suggesting that they are poised for activation in pSLE B cells (figure 3C).
Discussion
We are the first to investigate epigenomic changes in paediatric-onset SLE using ATACseq. In a cohort of 10 children with active, treatment-naive patients with pSLE, who were compared with 5 healthy children, we demonstrated differences in chromatin accessibility in peripheral blood B cells. There were 50.34% of the peaks in patients with pSLE located intergenic and distally from promoter sites, indicating that many transcription factors may be acting as distal enhancers to regulate transcription. The DAR from pSLE B cells were enriched for enhancer histone marks. Several transcription factor binding motifs were enriched within the DAR, indicating their involvement in regulating transcription of target genes. Fifty-five per cent of DAR located within SLE haplotypes appear to be within 1 kbp of genes involved in cell proliferation, activation of the adaptive immune response, activation of the innate immune response and cellular adhesion, for example, CCL4, IRF5, STAT4 and TNFAIP3.
Our data are similar to ATACseq data in adult SLE B cells: Scharer et al14 reported increased accessibility in a range of transcription factors involved in B cell activation and B cell differentiation. They also reported differential accessibility between SLE and healthy adult B cells in the STAT4 promoter, leading them to conclude that SLE B cells are epigenetically unique from healthy adult B cells. Our analysis of their raw ATACseq data demonstrated further epigenetic differences between paediatric and adult lupus B cells. Most notably, we found more regions of inaccessible chromatin in adult SLE B cells than those from pSLE, suggesting decreased levels of transcription of many associated genes. A larger proportion (65.2%) of the DAR in B cells from paediatric patients with lupus were located within SLE haplotypes compared with 39.7% in adult patients with lupus, suggesting that more regions of open chromatin in paediatric lupus B cells may be poised for transcription and contribute to disease pathogenesis. This is consistent with studies in adult autoimmunity that demonstrated enrichment of SNPs within loci associated with immune-mediated diseases located at specific binding sites, such as around STAT3, that are important in regulating transcription of proinflammatory genes.48
Limitations to our study include a small sample size and the use of total B cells in both patients with pSLE and healthy children. We were limited in the starting material for ATACseq and would not have had enough B cells in each group if we had attempted to perform bulk ATACseq in B cell subsets. However, we acknowledge the importance of studying the B cell subsets individually, as specific subsets have been shown to directly contribute to disease pathogenesis (eg, plasmablasts). Scharer et al14 demonstrated feasibility of performing bulk ATACseq in B cell subsets from adult patients with lupus; however, the amount of starting material to perform bulk ATACseq in each B cell subset would be prohibitive in patients with pSLE. Studies are currently underway by our group to determine feasibility of using single-cell ATACseq to evaluate B cell subsets from patients with pSLE. Single-cell ATACseq would be the appropriate next methodology to use as it is particularly useful for describing rare, transient populations.
Moreover, we acknowledge that our data would be strengthened by corresponding data from transcriptomes of pSLE B cells compared with B cells from healthy children. Here again, we were limited in our starting material for bulk ATACseq and were unable to perform RNA sequencing in the same samples from treatment-naive pSLE or healthy B cells. Single-cell RNA sequencing has been successfully performed to characterise peripheral blood mononuclear cells from paediatric patients with lupus,49 confirming feasibility. Genes important in cellular adhesion and the proinflammatory response, including genes contributing to the interferon gene signature, were found to be upregulated in paediatric patients with lupus. Several of these genes, including CCL4, interferon-induced protein 44 (IFI44), and rho-associated coiled-coil containing protein kinase 1 (ROCK1), were similarly found to reside in DAR described in our study, indicating that they are more accessible for transcription in our paediatric lupus cohort.
In addition, functional follow-up of identified disease-causing variants and their mechanisms of action remain challenging in paediatric autoimmune disease. More specifically, studies in adult autoimmune diseases have identified altered transcription factor binding to enhancers with specific disease-causing variants using in vitro and/or in vivo assays.48 50 51 Studies in T cells from adults with autoimmune disease have identified over 90% of causal variants to reside in non-coding regions of the genome, and only 10%–20% of identified variants appear to directly alter transcription factor binding motifs.52 Using more targeted methodologies such as clustered regularly interspaced short palindromic repeats (CRISPR) will allow us to further delineate and decipher disease-causing genetic variants in autoimmune diseases, such as pSLE.53 While beyond the scope of the current work, studies are underway in our laboratory to determine the optimal methodology to investigate genetic variants that cause this rare, paediatric autoimmune disease.
In conclusion, this is the first report describing chromatin architecture in children with pSLE. We demonstrate an epigenetically distinct profile in pSLE B cells when compared with those from healthy children, healthy adults and adults with SLE, indicating that pSLE B cells are predisposed for disease onset and development. Differences in chromatin accessibility between B cells in patients with pSLE, adult patients with SLE and healthy individuals, particularly in pathways controlling inflammation and activation of the immune response, suggest that transcriptional dysregulation of key players in B cell activation and differentiation plays an important role in the pathogenesis of pSLE.
Data availability statement
Data are available in a public, open access repository. ATACseq data sets generated and analysed for this manuscript are uploaded into Sequence Read Archive, per protocol, accession number PRJNA884097 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA884097). Adult ATACseq data set is publicly available at Sequence Read Archive, accession number PRJNA290920 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA290920).
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Northwell Health IRB protocol HS016-0017. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the patients and families for their participation in this study. This work would not have been possible without the bioinformatics expertise of Dr Frank Jenkins (1967-2021). We thank Andrea La Bella and Daniel Lulo for technical expertise and database management. We thank Drs Suhas Ganguli, Mei Ma, Roya Samuels, Minu George, Beth Gottlieb and Katherine Steigerwald for their clinical care.
References
Footnotes
Contributors JH-Y was involved in study conceptualisation and design, data collection and all aspects of data analysis. JH-Y wrote the first draft of the manuscript and performed revisions. KJ and SM performed all wet lab experiments including sample processing, B cell isolation and building libraries for ATACseq experiments and helped revise the manuscript. BAE and HW were involved in patient recruitment, sample procurement and helped draft the manuscript. BD was involved in study conceptualisation and design and helped revise the manuscript. JJ was involved in study conceptualisation, design and coordination and wrote the first draft of the manuscript with JH-Y. JH-Y accepts full responsibility for the conduct of the study as guarantor, had access to the data, and controlled the decision to publish. All authors read and approved the final manuscript.
Funding JH-Y is the recipient of a Scientist Development Award from the Rheumatology Research Foundation. This work was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001412 to the University at Buffalo.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.