Article Text

Abstract
This review describes eight ‘great ideas’ regarding bench-to-bedside considerations in systemic lupus erythematosus (SLE) presented at the second international LUPUS meeting in Quebec, September 2014. The topics included: correcting the impaired clearance of apoptotic fragments; optimisation of clinical trial design: the PERFECT (Pre Evaluation Reducing Frighteningly Elevated Coverable Targets) study; lipidomics and metabolomics in SLE; importance of the inflammasome; identification and treatment of asymptomatic autoimmunity: prevention of SLE; combining low doses of hydroxychloroquine and quinacrine for long-term maintenance therapy of SLE; reducing emergency room visits and the critical relevance of the autoantigen.
- Systemic Lupus Erythematosus
- Inflammation
- Autoimmunity
- Disease Activity
- Lupus Nephritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
For a second time, Drs Paul R. Fortin, John Esdaille, Matthew Liang, Peter E. Lipsky and Daniel J. Wallace hosted a specialised conference which brought together basic and clinical researchers all engaged in a common quest to significantly improve the lives of patients with systemic lupus erythematosus (SLE). The LUPUS 2014 meeting was held in Quebec on 22–24 September 2014. A clear intent of this meeting, in which timely science and medicine were reviewed, was to provoke lively discussion among the participants. To this end, the organisers pushed many of the invited speakers to take controversial stands on several state-of-the-art topics, from the importance (or not) of the interferon signature in therapeutic targeting to new frontiers in the microbiome. The tone of the meeting was set on opening night when the provocateurs, Drs Peter Lipsky and Paul Fortin, called upon several prewarned attendees to present their best great idea in 5 min illustrated by one slide. In the spirit of transparency and accelerating the pace of discovery in lupus, all participants agreed to share their original nugget. In keeping with the often repeated mantra of Dr Gerald Weissmann, “Don't tell me about your latest idea, show me your paper”, the ideas are presented herein.
Correcting the impaired clearance of apoptotic cell fragments
PC summarised the evidence for the generally accepted view that incomplete clearance of apoptotic cell fragments leads to self-immunisation with lupus autoantigens and may serve to initiate or perpetuate disease.1 Numerous animal models in which phagocytosis of apoptotic fragments is impaired due to absent receptors were discussed, andPC reviewed direct observational evidence in lupus of incompletely cleared apoptotic debris.2 The Mer tyrosine kinase is a key receptor for apoptotic debris. It recognises dying cells by binding to exposed phosphatidylserine residues through the intermediary molecules Protein S and Gas 6. PC suggested that cross-linking Mer might increase its efficiency in accomplishing apoptotic cell clearance by increasing its effective valence on the cell surface. He presented data showing that a recently developed monoclonal anti-mouse Mer increased phagocytosis of apoptotic lymphocytes. This was not true of all monoclonal anti-Mer antibodies, suggesting that the monoclonal antibody-binding site may be of importance in enhancement of clearance. These studies may provide the basis for further investigations of Mer as a target for enhancing clearance in autoimmune states such as lupus, reducing the amount of ongoing self-immunisation and decreasing autoimmunity.
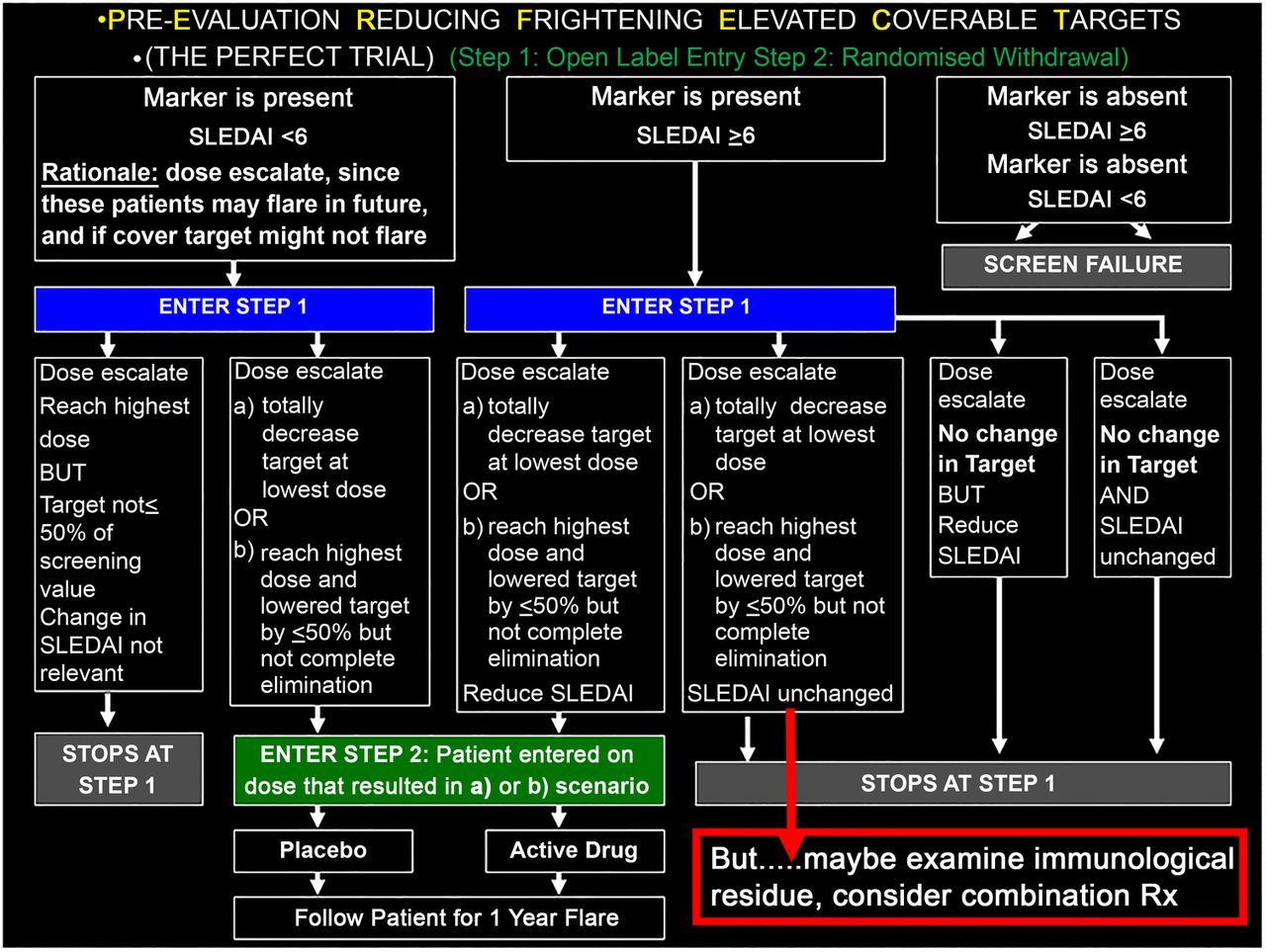
Optimisation of clinical trial design: the PERFECT (Pre Evaluation Reducing Frighteningly Elevated Coverable Targets) study
JPB submitted that there are many great ideas in thinking about SLE, but no great idea will fit all SLE—because SLE is heterogeneous. Perhaps the greatest idea is to simply acknowledge this fact, and accept its profound consequences for the pharmaceutical industry, that one targeted treatment will not work optimally for all patients, one dose will not work for even all of the appropriate patients and some patients may require addressing more than one immunological pathway. To address this (perhaps obvious but nevertheless iconoclastic notion may not be entirely congruent with Federal Drug Administration (FDA) guidelines for treatment development—http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072063.pdf), JPB partnered with JTM and proposed a trial design which considers individuality by focusing on clinical activity that is accompanied by activation of a treatment-specific, pharmacodynamic target. The PERFECT (Pre Evaluation Reducing Frighteningly Elevated Coverable Targets) trial begins with an open–label, dose escalation study in patients known to have dysregulation in a pathway targeted by a given biologic to initially determine the optimal dose for target coverage. This achieves two goals: it assures a reduction in the desired target has been made at an optimal dose for each patient (ideally total reduction but acknowledging 50% might be acceptable if that is the ceiling using the highest safely tolerated dose), and eliminates patients who have <50% reduction since the drug is not likely to have benefit. Those achieving target reduction are then blindly randomised to remain on their optimal dose or be withdrawn with the primary endpoint being evaluation of flares (summarised in figure 1). For patients who achieve target coverage but who don't have a clinical response, exploration of additional active pathways or immunological feedback loops in response to the therapy might suggest logical combination treatments for these individuals.
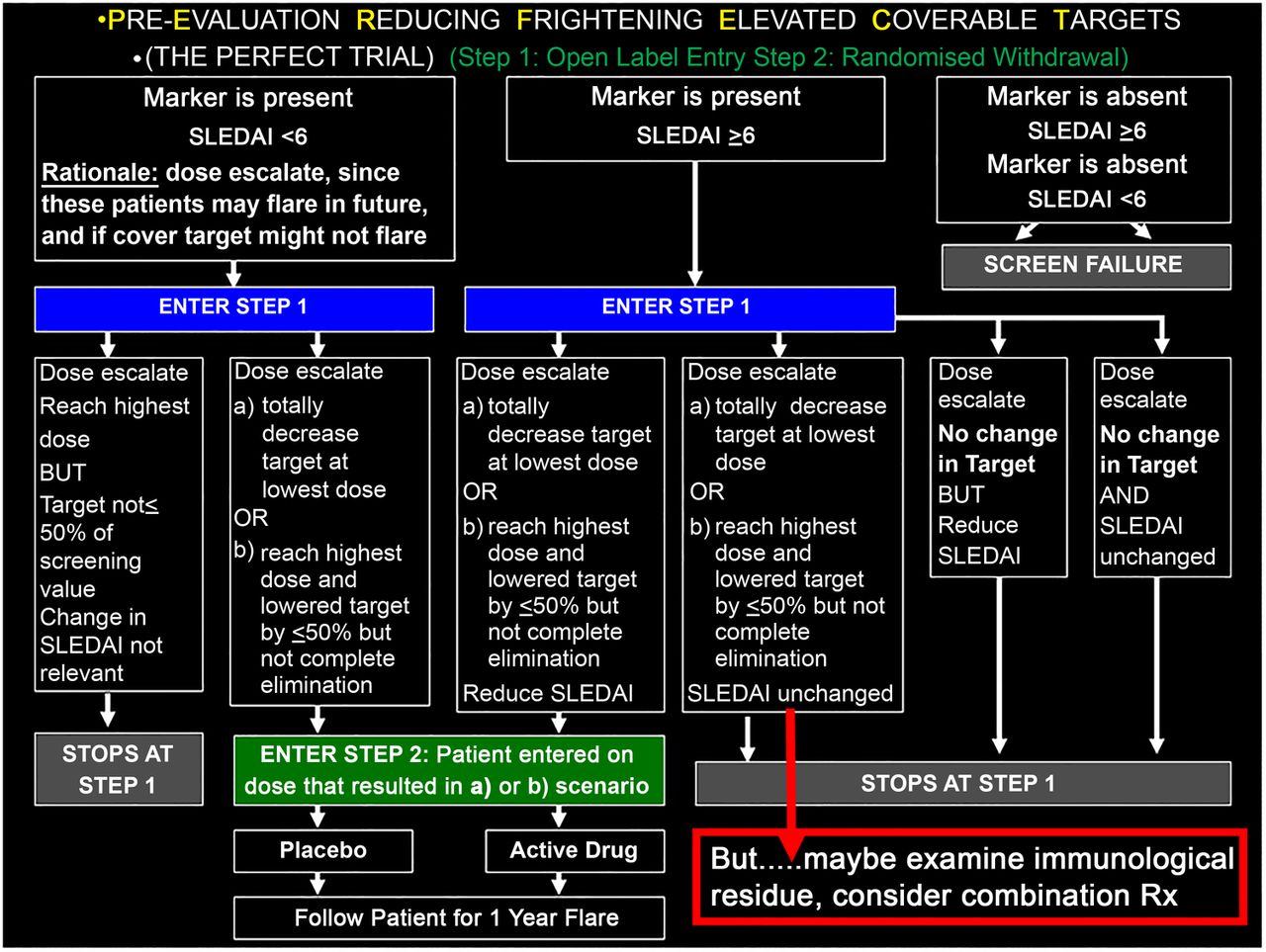
Proposal for the PERFECT Trial.
Lipidomics and metabolomics in SLE
GG focused on the growing acknowledgement that agnostic omics type screens have the potential for defining new pathways of pathogenesis in SLE. Proteomics, especially in defining urinary proteins in lupus nephritis, is an active ongoing field of research.3 New omics assessments are now possible due to the development of novel technologies. These include lipidomics and metabolomics at the cellular, organ and systemic level. Lipids play a key role in cell signalling and cell death. These lipid pathways are targetable and thus are potential therapeutic targets in SLE. Lipidomic assessments in human and murine lupus are shown in figure 2.4 The left panels show differences in lipid expression in patients with SLE compared with controls. The top panel A demonstrates that there is significant elevation of lactosyl ceramides in the urine of patients with lupus nephritis compared with urine from patients with lupus without renal disease and controls. (Panel B demonstrates that serum levels of this same ceramide, in contrast to marked differences in urinary levels, are similar in patients with or without nephritis, suggesting that the urinary levels are representative of renal production of the ceramide.) The bottom panel demonstrates staining for lactosyl ceramide in the kidney of a control, one patient with Class III lupus nephritis and one with Class IV lupus nephritis. Thus, one can demonstrate local expression of this lactosyl ceramide in the glomerulus of patients with active lupus nephritis.

Lipidomic assessments in human and murine lupus.
The top right panel shows mass spectrometry assessments of different ceramides in the kidneys of control Black 6 mice (B6) and kidneys from a lupus mouse MRL/lpr (lupus nephritis). The hotter the colour, the higher is the level of expression. The bottom panels (F) demonstrate that there is differential expression of ceramides in the cortex versus medulla depending on chain length. The bottom right panel G demonstrates similar patterns of expression of some lipids, while others are uniquely expressed in the lupus (MRL/lpr) kidney.
In summary, lipidomic assessment of patients with lupus indicates clear differences from controls. Further assessment is needed to fully characterise these abnormalities and the specific pathways/enzymes involved. Therapeutic targeting strategies could then be developed that potentially treat acute disease but also possibly prevent chronic deterioration of renal function in patients with lupus nephritis.
Importance of the inflammasome
MK emphasised that while autoimmune diseases and autoinflammatory diseases have traditionally been considered separate and distinct entities, recent evidence indicates that activation of the inflammasome machinery may play important roles in lupus immune dysregulation and lupus-associated organ damage. Furthermore, although the dogma has been that abnormalities in apoptotic clearance of apoptotic material are key mechanisms leading to externalisation of modified autoantigens, it is important to consider the role that more proinflammatory types of cell death play in autoantigen modification, induction of autoimmune responses and tissue damage in SLE. Among various forms of cell death, pyroptosis deserves particular consideration as a proinflammatory mechanism. This cell death can lead to profound propagation of cytokine production and tissue damage through a novel mechanism of cell–cell communication through ‘jumping’ components of the inflammasome machinery.5 Given that a subset of individuals with autoimmune disorders develop adaptive immune responses directed at these inflammasome components and that these antibodies have opsonising and proinflammatory properties, better characterising the role of the inflammasome in mediating systemic autoimmunity is warranted.
Identification and treatment of asymptomatic autoimmunity: prevention of SLE
JJ pointed out that by the time patients with SLE meet lupus classification criteria, they often have irreversible organ inflammation and associated damage. Over time, end-organ damage accumulates, leading to extensive morbidity and early mortality. In an effort to suppress inflammation and minimise organ damage, patients are oftentimes treated with significant immunosuppressive drugs, such as mycophenolate mofetil, azathioprine and corticosteroids, which present additional risks of infection, liver damage, osteonecrosis, osteoporosis, cataracts and accelerated atherosclerosis. Moreover, these therapies fail to selectively address the underlying cause of chronic inflammation in SLE: pervasive, directed immune dysregulation.
Recent and ongoing studies suggest that immune dysregulation begins well before SLE is clinically apparent. In one model of SLE pathogenesis, in genetically predisposed individuals, select environmental triggers stimulate autoantibody production, initiating a period of benign autoimmunity (figure 3). Some individuals remain in this period of benign autoimmunity indefinitely, while others transition to pathogenic autoimmunity. This suggests that after the appearance of autoantibodies, a secondary insult such as inappropriate inflammatory cytokine production leads to increased autoantibody formation and further activation of T and B lymphocytes. With these developments, the patient enters a period of preclinical pathogenic immunity. As antibody targets diversify, cytokine production increases and tissue damage accumulates, the early signs of SLE become clinically apparent. The patient finally meets the criteria for SLE classification, after irreversible damage has occurred and well after the onset of pathogenic autoimmunity.
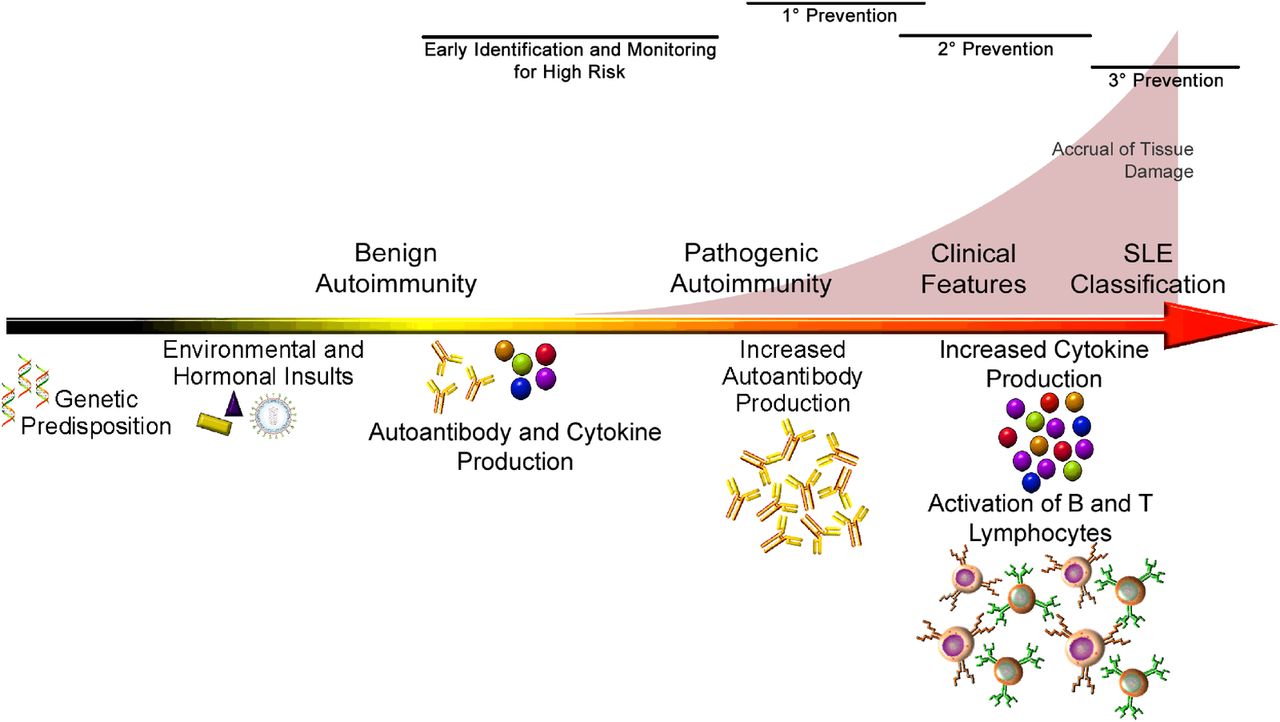
Proposed working model of SLE pathogenesis and progression from clinically asymptomatic to overt disease.
Thus, by the time we begin to ‘treat lupus’, it may already be too late to preserve the patient's health. In contrast, preventing SLE could eliminate the risk of irreversible organ damage and might even allow the patient's immunoregulatory mechanisms to re-establish normal homeostasis. Indeed, this approach is already being pursued for other autoimmune diseases, including juvenile diabetes (type 1) and rheumatoid arthritis. To make this leap with SLE, it should be determined who needs early intervention and how the pathogenic process can be safely interrupted. Studies from longitudinal data and samples from preclinical SLE collections have shown that autoantibodies precede clinical disease in linked subsets6–8 and increase in concentration immediately preceding classification,9 ,10 select symptoms are common before disease classification6 and hydroxychloroquine use may slow the accrual of new autospecificities and delay onset of clinical disease.11 Ongoing follow-up studies in blood relatives of patients with SLE, as well as in longitudinal cohorts of individuals who later develop lupus,12 should provide the information necessary to identify the high-risk individuals who would be candidates for prevention trials. The published literature and the strong benefit:risk ratio make hydroxychloroquine an attractive choice for the initial drug to be tried in lupus prevention studies. Moreover, a better understanding of the early mechanisms of disease transition may lead to targeted-immune therapies, such as anti-BLyS or induced regulatory cell therapy that could be used in short-term interventions to prevent transition in high-risk individuals.
A proposal to combine low doses of hydroxychloroquine and quinacrine for long-term maintenance therapy of SLE
WJMC proposed a novel method for long-term management of SLE. Multiple benefits of antimalarial therapy for lupus are now widely recognised in addition to the traditional role of achieving control of minor disease manifestations. These include prevention of both major and minor disease flares, reduction of mortality in multiple large cohorts, reduced accrual of damage, lower risk of developing nephritis and improved outcome of treatment of nephritis. Favourable effects on cardiovascular risk have been reported in studies of hydroxychloroquine, chloroquine and quinacrine, including improvement of dyslipidemias, prevention of diabetes in rheumatoid arthritis, improvement of glucose control in diabetes and anti-thrombotic effects in lupus and anti-phospholipid syndromes.
The promise of hydroxychloroquine to simultaneously prevent disease flares and reduce cardiovascular risk encourages long-term use. Unfortunately, long-term follow-up of patients treated with hydroxychloroquine, as documented in case series13 and as reflected by newer guidelines for prevention of ocular toxicity14 suggests that doses of hydroxychloroquine previously felt to be safe (eg, 200 mg/day for an average-sized patient) are associated with significantly increased risk of ocular toxicity during prolonged administration. Hence, it is reasonable to ask whether it would be advantageous to combine therapy with a lower dose of hydroxychloroquine (50 or 100 mg/day) with a low dose of quinacrine, since, in contrast to hydroxychloroquine and chloroquine, quinacrine has not demonstrated retinal toxicity. Millions of American soldiers used quinacrine for antimalarial prophylaxis during World War II without ocular toxicity15 and a later detailed evaluation of 26 patients who received 18–102 g of quinacrine showed no retinal toxicity.16
A review of quinacrine therapy used to treat more than 700 patients in the pre-steroid era suggests that it is effective in both cutaneous and systemic diseases.15 Combination therapy with quinacrine, hydroxychloroquine and/or chloroquine for cutaneous or systemic lupus has been reported in several series, most notably the1959 report in the NEJM of use of ‘Triquin’ (hydroxychloroquine 50 mg, chloroquine 65 mg and quinacrine 25 mg).17 Triquin was FDA approved and widely used from 1959 to 1972 when the FDA withdrew approval of this as part of a campaign against combination drugs. Combination antimalarial therapy for lupus remains widely used in dermatology.18 High doses of quinacrine have been associated with central nervous system activation, including seizures and hypomania, as well as yellow discolouration of the skin; use of a low dose, for example, 25 mg/day would be expected to minimise these problems.
Even if quinacrine is safe for the retina as a single agent, is it certain that co-administration with hydroxychloroquine does not increase the toxicity of the latter? This is unknown but it seems unlikely that the additional risk would be as great as that of using an increased dose of hydroxychloroquine alone. In addition, the ‘metabolic’ benefits noted above are less well studied with quinacrine.
Reducing emergency room visits
SB focused her great idea on decreasing emergency room (ER) visits, which are common events for patients with SLE. One study from California showed that among a group of 807 patients with SLE, only a small number (n=78, 10%) accounted for the majority of the group's ER visits19 These frequent users had greater SLE disease activity and worse general health and were less likely to be employed and/or to have a college degree. Moreover, a recent assessment of patients with lupus nephritis, based on Medicare data from the USA, suggested that a proportion of these patients consistently use the ER for regular care,20 which is not only inefficient and costly, but also points to a need to address problems with medical access for some of our most vulnerable patients with SLE. This phenomenon has implications not only for cost but also for the quality of care received. In a survey of ER physicians’ attitudes towards ‘frequent users’ of ER care, 91% of respondents called frequent users ‘a problem’, 77% admitted a bias against frequent users and 59% said they have less empathy for these patients than for other patients.21 Attempts to address the phenomena of frequent ER visits in the general population have had “…mixed success at best”,22 which is believed to be due in part to the heterogeneity of the population of frequent users, some of whom have important, complex medical problems, like SLE. Authorities have called for more research, pointing out that frequent ER visitors likely represent a psychosocially vulnerable group.23
What further research, then, could help us understand the problem of frequent ER visits in patients with SLE, and how we can optimise care for these patients? First, attempts should be made to more comprehensively (and across many different jurisdictions) quantify the burden of frequent ER visits in SLE, both in terms of direct and indirect costs. Second, quantitative research, such as focus groups, including both patients with SLE and care providers (lupus specialists, family physicians and ER physicians), could clarify the issues surrounding frequent ER use by patients with SLE. This should ultimately help identify barriers to optimal care for patients with SLE, and potential solutions that might be of use in addressing the problem of frequent ER use by a vulnerable subset of our patients with SLE.
It's the antigen, stupid
KE discussed the critical relevance of the autoantigen in SLE pathogenesis. “It’s the economy, stupid” is a slight variation of the phrase “The economy, stupid” which James Carville had coined as a campaign strategist of Bill Clinton’s successful 1992 presidential campaign against sitting President George H. W. Bush.24
Patients with SLE have high titres of autoantibodies which have been estimated to reach up to mg/mL concentrations in serum.25 Yet many of these patients with high autoantibody concentrations are in remission—why aren't they sick? The idea presented by KE is that the limiting factor is availability of antigen. This explains why cell death from exposure to ultraviolet (UV) light precipitates both local disease and flare of systemic disease since availability of antigen allows immune complexes to form and provoke inflammation in patients with SLE.
KE reasoned using coeliac disease as a model where both the trigger (gluten) and drivers (activated T and B memory cells) lead to production of autoantibodies (in this case targeted to transglutaminase 2) (figure 4, left panel). The remarkable observation is if the trigger—the offending external antigen—is removed, the disease goes into remission. Note that survival of memory T and B cells renders patients exquisitely sensitive to exacerbations of disease upon re-exposure to antigen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Celiac disease as a model for SLE. In celiac disease, both the trigger (gluten) and drivers (activated T and B memory cells) lead to production of autoantibodies (in this case targeted to transglutaminase 2) (left panel). If the offending external antigen is removed, the disease goes into remission but persistence of memory T and B cells renders patients exquisitely sensitive to exacerbations of disease upon re-exposure to antigen. In SLE (right panel) the dominant site of antigenic stimulation is proposed to be skin rather than gut. Here, UV light acts as the trigger. In contrast to cytotoxic CD8 T cells in celiac, the key driver in SLE is memory B cells but T cells may also play a role. (Figure modified from Ludvig et al. Nat Rev Immunol 2013;13:294–303)
So, while the site of antigenic stimulation may be different in SLE, the idea he proposed is that in SLE, antigen is the key limiting element and that UV light in SLE acts like gluten in coeliac disease (figure 4, right panel). In contrast to CD8T cells in coeliac, he proposed that a key driver is memory B cells.
Pharmaceutical companies have spent hundreds of millions of dollars on trying to get rid of antibodies (B cell depletion or modulation). Unlike gluten in coeliac disease, we cannot simply get rid of the trigger (sunshine) in SLE, but strategies to limit or degrade antigen, especially the nucleic acid component, may yet prove a new approach to treatment of this disease.
Acknowledgments
The authors thank Dr Paul Fortin (Chair) and Drs John Esdaile, Matthew Liang, Peter E. Lipsky and Daniel J. Wallace (Committee Members) for organising the Lupus 2014 meeting in Quebec.
References
Footnotes
Contributors All authors contributed equally to the writing of this manuscript.
Funding PC has received research funding from Janssen Laboratories, LLC. GG is funded by VA Medical Research Service, Ralph H. Johnson VAMC.JJ is supported by NIH grants U19AI082714, U01AI101934, U54GM104938, P30GM103510, P30AR053483.
Competing interests Keith Elkon is co-founder of Resolve Therapeutics.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement No additional data are available.