Article Text

Abstract
Objective Defective or inefficient DNA double-strand break (DSB) repair results in failure to preserve genomic integrity leading to apoptotic cell death, a hallmark of systemic lupus erythematosus (SLE). Compelling evidence linked environmental factors that increase oxidative stress with SLE risk and the formation of DSBs. In this study, we sought to further explore genotoxic stress sensitivity in SLE by investigating DSB accumulation as a marker linking the effect of environmental stressors and the chromatin microenvironment.
Methods DSBs were quantified in peripheral blood mononuclear cell subsets from patients with SLE, healthy controls, and patients with rheumatoid arthritis (RA) by measuring phosphorylated H2AX (phospho-H2AX) levels with flow cytometry. Phospho-H2AX levels were assessed in G0/G1, S and G2 cell-cycle phases using propidium iodide staining, and after oxidative stress using 0.5 µM hydrogen peroxide exposure for 0, 2, 5, 10, 30 and 60 min.
Results DSB levels were significantly increased in CD4+ T cells, CD8+ T cells and monocytes in SLE compared with healthy controls (p=2.16×10−4, 1.68×10−3 and 4.74×10−3, respectively) and RA (p=1.05×10−3, 1.78×10−3 and 2.43×10−2, respectively). This increase in DSBs in SLE was independent of the cell-cycle phase, and correlated with disease activity. In CD4+ T cells, CD8+ T cells and monocytes, oxidative stress exposure induced significantly higher DSB accumulation in SLE compared with healthy controls (60 min; p=1.64×10−6, 8.11×10−7 and 2.04×10−3, respectively).
Conclusions Our data indicate that SLE T cells and monocytes have increased baseline DSB levels and an increased sensitivity to acquiring DSBs in response to oxidative stress. Although the mechanism underlying DSB sensitivity in SLE requires further investigation, accumulation of DSB may serve a biomarker for disease activity in SLE and help explain increased apoptotic cell accumulation in this disease.
- Double strand breaks
- Oxidative stress
- H2AX phosphorylation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Double-strand DNA breaks are increased in lupus T cells and monocytes independent of the cell-cycle phase.
Double-strand DNA breaks accumulation, assessed using flow cytometry quantifying phosphorylated histone H2AX, correlates with disease activity in lupus.
Lupus T cells and monocytes are more sensitive to the genotoxic effect of oxidative stress compared with healthy controls.
Impaired double-strand DNA break repair in lupus might contribute to increased apoptosis, a hallmark of the disease.
Introduction
Systemic lupus erythematosus (SLE or lupus) is a complex disease characterised by autoantibody production and the involvement of multiple organ systems. Although the pathogenesis of SLE remains unclear, there is a well-established genetic predisposition to the disease with ∼60 genetic risk loci confirmed to date.1–3 Several environmental factors are also known to increase the risk for SLE, including silica dust exposure, mercury, ionising radiation, ultraviolet light and oxidative stress.4 The overwhelmingly unifying factor among almost all environmental exposures known to induce SLE is the potential to be genotoxic by producing reactive oxygen intermediates and double-strand DNA breaks.
Double-strand breaks (DSBs) represent a severe form of DNA damage that can cause structural instability, genetic mutations and cell apoptosis if the damage is not effectively repaired.5 ,6 Defective DSB repair has recently been associated with SLE with evidence for inefficient or delayed repair of DSBs in Epstein–Barr virus (EBV)-transformed B-cell lines derived from patients with paediatric SLE.7 In addition, both intrinsic and genotoxic stress-induced DSB accumulation were shown to be increased in peripheral blood mononuclear cells (PBMCs) from patients with lupus nephritis.8
In this study, we sought to further investigate the relationship between SLE and DSB accumulation through in-depth analyses in primary CD4+ T cells, CD8+ T cells and monocytes. DSB levels were assessed by quantifying phosphorylation of serine 139 on H2AX (phospho-H2AX), a well-established DSB biomarker, which is involved in amplifying the DNA damage response signalling cascade9 ,10 and is one of the earliest, most robust cellular responses to DNA damage.11 ,12 Disease-associated changes in phospho-H2AX levels were evaluated in CD4+ T cells, CD8+ T cells and monocytes from patients with SLE compared with healthy controls and patients with rheumatoid arthritis (RA) as disease controls. We also examined the relationship between phospho-H2AX levels and SLE disease activity. In each of the three cell subsets, we investigated the effectiveness of DSB repair in SLE by assessing phospo-H2AX levels in response to oxidative stress.
Materials and methods
Patients with SLE and matched controls
Our study group comprised a total of 18 female patients with SLE, 15 female healthy controls and eight female patients with seropositive RA. Each patient with SLE fulfilled at least four of the American College of Rheumatology classification criteria for SLE.13 In addition, none of the recruited patients had prior exposure to either cyclophosphamide or calcineurin inhibitors. All participants were recruited from the University of Michigan rheumatology clinics, and signed an informed consent approved by our institutional review board. The disease activity of each patient was assessed using the SLE disease activity index (SLEDAI),14 by trained individuals in our Michigan Lupus Cohort. A summary of the demographic information, medications and SLEDAI scores for each patient included in this study is shown in table 1.
Demographic and medication information are shown for patients with SLE and healthy matched controls included in the study
Peripheral blood mononuclear cell isolation
Fresh peripheral blood samples (80 mL) were collected from each study participant. Ficoll-Paque density gradient centrifugation (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was used to isolate PBMCs. After isolation, PBMCs were either stained for direct analysis by flow cytometry or used for the hydrogen peroxide treatment experiments prior to analysis.
Treatment of cells with hydrogen peroxide
PBMCs were incubated for 60 min in Roswell Park Memorial Institute medium (Lonza, Walkersville, Maryland, USA) supplemented with 10% of donor's serum, 10 000 U/mL penicillin and 10 mg/mL streptomycin (Sigma-Aldrich, St. Louis, Missouri, USA). During incubation, the cells were exposed to 0.5 µM H2O2 (Sigma-Aldrich, St. Louis, Missouri, USA) for 0, 2, 5, 10, 30 or 60 min at 37°C in a 5% CO2 atmosphere.
Flow cytometry and antibody staining
PBMCs were stained with Pacific Blue anti-human CD3, APC anti-human CD4, PE anti-human CD8a and APC/Cy7 anti-human CD14 (Bio Legend, San Diego, California, USA). The stained cells were then fixed and permeabilised using the Nuclear Factor Fixation and Permeabilization Buffer Set (BioLegend, San Diego, California, USA) as per the manufacturer's instructions. Intracellular staining was then performed using fluorescein isothiocyanate anti-phosphorylated (ser139) H2AX (clone 2F3; BioLegend). For the cell-cycle experiments, the PBMCs were additionally treated with 20 μg/mL RNase A (Sigma-Aldrich, St. Louis, Missouri, USA), and then stained with 40 μg/mL propidium iodide (BioLegend) as per manufacturer's instructions for 1 hour prior to analysis. Flow cytometry analysis was then performed using a MoFlow Astrios Flow Cytometer and Summit software V.6.2.3 (Beckman Coulter, Miami, Florida, USA). Phospho-H2AX levels are reported as median fluorescence intensity (MFI) values.
Statistical analysis
Phospho-H2AX levels were compared between SLE, healthy control and RA groups using unpaired Student's t tests that assumed equal variance between groups. The relationship between phospho-H2AX levels and SLEDAI was assessed by Pearson correlation analyses. For H2O2 exposure time-course assays, phospho-H2AX levels were compared in each cell subset by multiple t tests that assumed equal variance, which were corrected for multiple testing using the Holm–Sidak method. Results were considered significant with a p<0.05, and all statistical analyses were performed using GraphPad Prism software V.6.07 (San Diego, California, USA).
Results
Baseline DSB levels are increased in patients with SLE
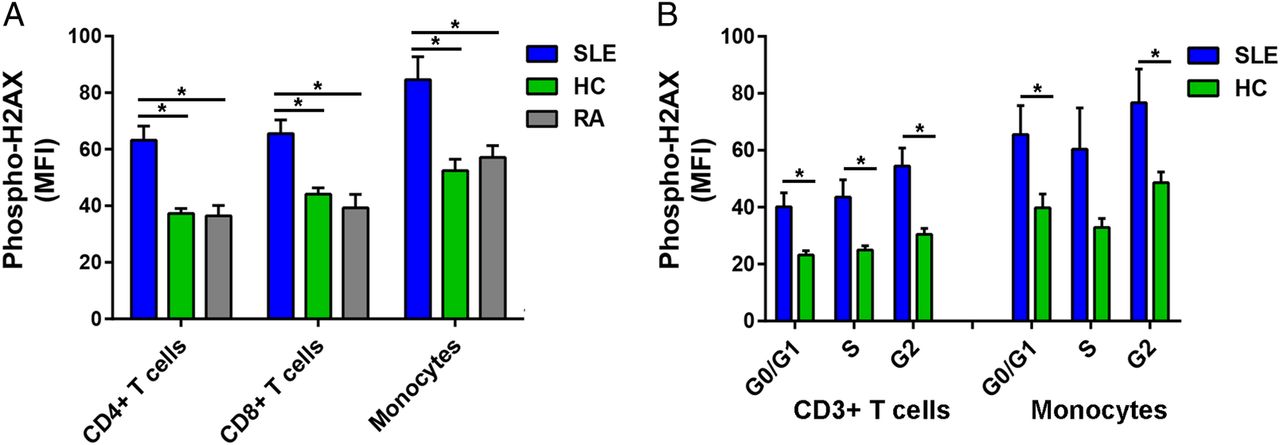
We examined DSB accumulation in 14 patients with SLE, 10 healthy controls and eight patients with RA by measuring levels of phospho-H2AX, a rapid and sensitive marker for DSBs.15 Phospho-H2AX levels were measured in freshly isolated primary CD4+ T cells, CD8+ T cells and monocytes by flow cytometry. For each of the three immune cell subsets, our results showed significantly higher levels of phospho-H2AX in SLE than in either healthy controls (average MFI; CD4+ T cells: 62.23 vs 37.28, CD8+ T cells: 65.56 vs 44.10, Monocytes: 84.60 vs 52.49; p=2.16×10−4, 1.68×10−3 and 4.74×10−3, respectively) or RA (average MFI; CD4+ T cells: 62.23 vs 36.45, CD8+ T cells: 65.56 vs 39.29, monocytes: 84.60 vs 57.11; p=1.05×10−3, 1.78×10−3 and 2.43×10−2, respectively; figure 1A).
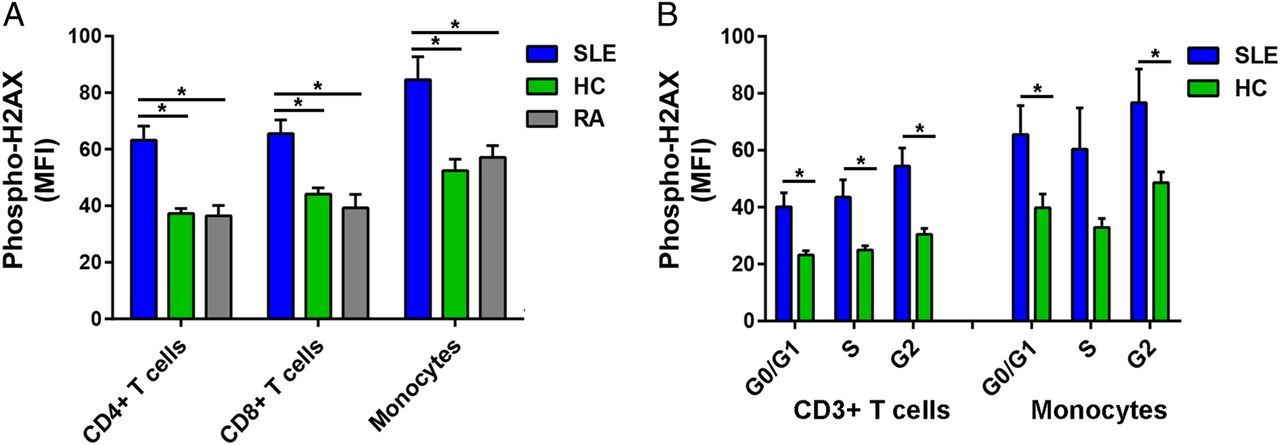
(A) Phospho-H2AX levels are significantly increased in CD4+ T cells, CD8+ T cells and monocytes from SLE compared with those from healthy controls (p=2.16×10−4, 1.68×10−3 and 4.74×10−3, respectively) and RA (p=1.05×10−3, 1.78×10−3 and 2.43×10−2, respectively). (B) Phospho-H2AX levels at G0/G1, S and G2 cell-cycle phases are significantly increased in CD3+ T cells in SLE compared with healthy controls (p=5.41×10−3, 9.71×10−3 and 2.86×10−3, respectively). In monocytes, there was a significant difference at G0/G1 and G2 cell-cycle phases (p=3.85×10−2 and 3.90×10−2, respectively), but the difference did not reach statistical significance at S phase (p=8.38×10−2). Phospho-H2AX levels were measured by flow cytometry, and are provided as MFIs (mean±SEM). HC, healthy controls; MFI, median fluorescence intensity; Phospho-H2AX, phosphorylated H2AX; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
We then determined if the increased phospho-H2AX levels in SLE were independent of differences in cell-cycle phases. We measured phospho-H2AX levels at G0/G1, S and G2 cell-cycle phases in CD3+ T cells and monocytes that were isolated from a subset of eight patients with SLE and eight healthy controls. In SLE compared with healthy controls, there were significantly increased levels of phospho-H2AX in CD3+ T cells at each of the three cell-cycle phases (average MFI; G0/G1: 40.10 vs 23.18, S: 43.57 vs 24.99, G2: 54.50 vs 30.44; p=5.41×10−3, 9.71×10−3 and 2.86×10−3, respectively; figure 1B) and in monocytes at G0/G1 and G2 (average MFI; G0/G1: 65.53 vs 39.76, G2: 76.75 vs 48.60; p=3.85×10−2 and 3.90×10−2, respectively; figure 1B). Together, these results demonstrate that patients with SLE exhibit significantly increased phospho-H2AX levels in CD4+ T cells, CD8+ T cells and monocytes. Furthermore, we show that this SLE-associated difference is independent of cell-cycle phase.
DSB accumulation correlates with SLE disease activity
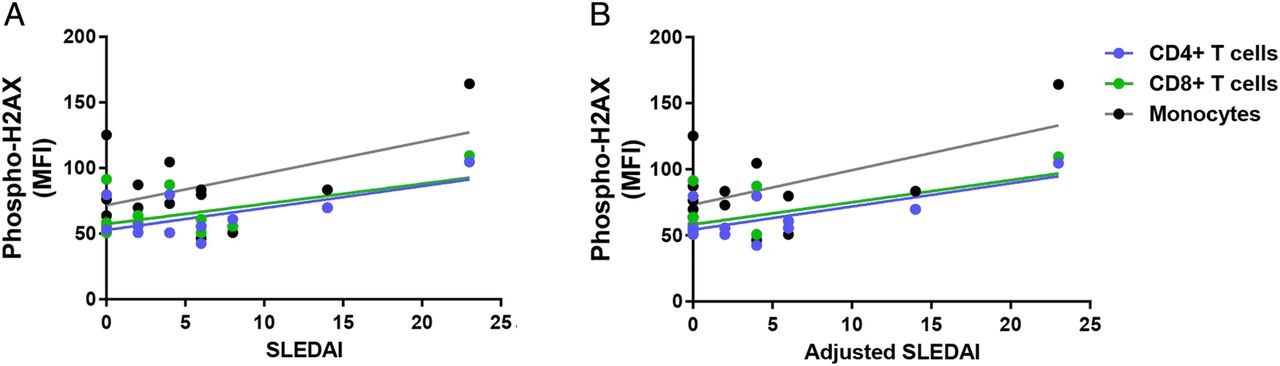
We investigated the relationship between the accumulation of DSBs and disease activity in patients with SLE. Our results showed a significant positive correlation between SLEDAI and phospho-H2AX levels in CD4+ T cells and CD8+ T cells (CD4+ T cells: r2=0.39, p=2.88×10−2; CD8+ T cells: r2=0.30, p=4.20×10−2; figure 2A). In addition, there was a significant positive correlation in CD4+ T cells, CD8+ T cells and monocytes when SLEDAI scores are adjusted to exclude criteria for low complement levels and increased anti-dsDNA antibodies (CD4+ T cells: r2=0.49, p=1.14×10−2; CD8+ T cells: r2=0.39, p=1.79×10−2; monocytes: r2=0.32, p=3.38×10−2; figure 2B). We then examined whether disease activity correlates with the accumulation of DSBs in different cell-cycle phases in a subset of eight patients with SLE. We showed significant positive correlations between SLEDAI and phospho-H2AX levels in CD3+ T cells (G0/G1: r2=0.77, p=4.30×10−3; S: r2=0.84, p=1.45×10−3; G2: r2=0.56, p=3.19×10−2; figure 3A) and in monocytes (G0/G1: r2=0.79, p=3.16×10−3; S: r2=0.77, p=4.39×10−3; G2: r2=0.54, p=3.87×10−2; figure 3A). Similarly, we showed significant positive correlations between the adjusted SLEDAI and phospho-H2AX levels in SLE CD3+ T cells (G0/G1: r2=0.74, p=6.01×10−3; S: r2=0.82, p=1.80×10−3; G2: r2=0.51, p=4.57×10−2; figure 3B) and monocytes (G0/G1: r2=0.77, p=4.44×10−3; S: r2=0.76, p=4.85×10−3; G2: r2=0.50, p=4.99×10−2; figure 3B).
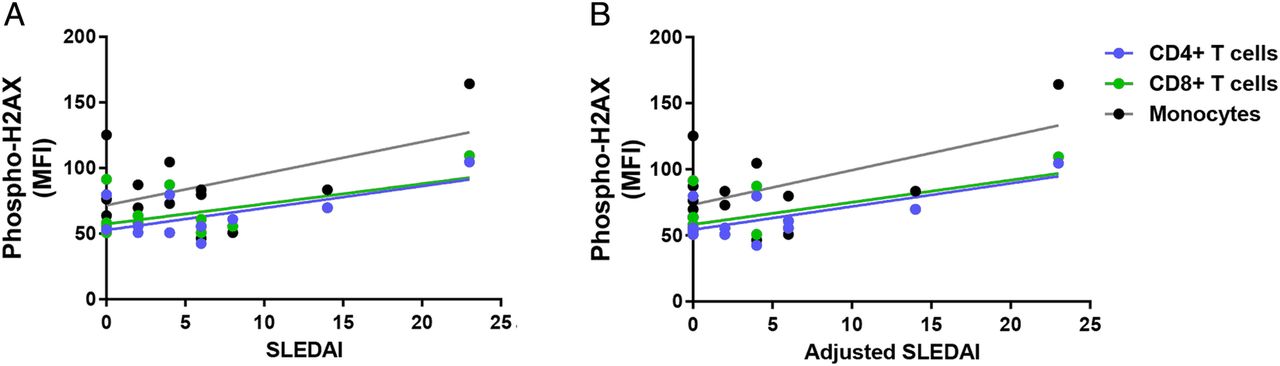
Correlation between disease activity and phospho-H2AX levels in primary CD4+ T cells, CD8+ T cells and monocytes from patients with SLE (n=12, 14 and 14, respectively). (A) SLEDAI scores were positively correlated with phospho-H2AX levels in primary CD4+ T cells and CD8+ T cells from patients with SLE (p=2.88×10−2and 4.20×10−2, respectively), but statistical significance was not reached in monocytes (p=6.24×10−2). (B) Adjusted SLEDAI scores, which excluded scoring criteria for low complement levels and increased anti-dsDNA antibodies, were positively correlated with phospho-H2AX levels in SLE CD4+ T cells, CD8+ T cells and monocytes (p=1.14×10−2, 1.79×10−2 and 3.38×10−2, respectively). Phospho-H2AX levels were measured by flow cytometry, and are provided as MFIs (mean±SEM). MFI, median fluorescence intensity; phospho-H2AX, phosphorylated H2AX; SLE, systemic lupus erythematosus; SLEDAI, SLE disease activity index.

Correlation between disease activity and phospho-H2AX levels at G0/G1, S and G2 cell-cycle phases in primary CD3+ T cells and monocytes from patients with SLE (n=8). (A) SLE disease activity index (SLEDAI) scores were positively correlated with phospho-H2AX levels at G0/G1, S and G2 cell-cycle phases in primary CD3+ T cells (p=4.30×10−3, 1.45×10−3 and 3.19×10−2, respectively) and monocytes (p=3.16×10−3, 4.39×10−3 and 3.87×10−2, respectively) from patients with SLE. (B) Adjusted SLEDAI scores, which excluded scoring criteria for low complement levels and increased anti-dsDNA antibodies, were positively correlated with phospho-H2AX levels at G0/G1, S and G2 cell-cycle phases in primary CD3+ T cells (p=6.01×10−3, 1.80×10−3 and 4.57×10−2, respectively) and monocytes (p=4.44×10−3, 4.85×10−3 and 4.99×10−2, respectively) from patients with SLE. Phospho-H2AX levels were measured by flow cytometry, and are provided as MFIs (mean±SEM). MFI, median fluorescence intensity; phospho-H2AX, phosphorylated H2AX; SLE, systemic lupus erythematosus; SLEDAI, SLE disease activity index.
Oxidative stress from hydrogen peroxide increases DSB levels more rapidly in patients with SLE compared with healthy controls
We tested whether patients with SLE have increased sensitivity to DSB accumulation in response to oxidative stress. In six patients with SLE and in six healthy age- and sex-matched controls, PBMC samples were isolated and treated with 0.5 µM hydrogen peroxide (H2O2) for 0, 2, 5, 10, 30 or 60 min. We used 0.5 µM H2O2 as this was the lowest concentration we tested in a preliminary experiment that increased phospho-H2AX levels above baseline. The exposure time points of 0, 2, 5, 10, 30 or 60 min were used to examine the trend between the initial increase of phospho-H2AX, which occurs rapidly following DNA damage, as well as the highest phospho-H2AX levels, which are reached 30–60 min after the damage.16 DSB accumulation in SLE and healthy control samples was detected in CD4+ T cells, CD8+ T cells and monocytes by measuring phospho-H2AX levels in each cell type by flow cytometry. Our data show significantly increased phospho-H2AX levels in SLE CD4+ and CD8+ T cells at 30 min (average MFI; CD4+ T cells: 893.16 vs 471.59, CD8+ T cells: 888.50 vs 435.23; p=2.51×10−3 and 3.32×10−3, respectively; figure 4A, B), and significantly increased phospho-H2AX levels in all three cell types at 60 min (average MFI; CD4+ T cells: 1176.30 vs 465.65, CD8+ T cells: 1233.01 vs 417.13, and monocytes: 421.87 vs 80.58; p=1.64×10−6, 8.11×10−7 and 2.04×10−3, respectively; figure 4A–C). It is also worth noting that phospho-H2AX levels decreased at the 60 min exposure time in each control cell type, whereas the levels continued to increase in each SLE cell type. Interestingly, monocytes from healthy controls showed limited change from baseline phospho-H2AX levels at each H2O2 exposure time point, suggesting a relative resistance in monocytes to accumulating DSBs compared with autologous CD4+ or CD8+ T cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phospho-H2AX levels in CD4+ T cells, CD8+ T cells and monocytes (A, B and C, respectively) are compared between patients with SLE and healthy controls in response to hydrogen peroxide exposure (0.5 µM) at 0, 2, 5, 10, 30 and 60 min (n=6). Phospho-H2AX levels were measured by flow cytometry, and are provided as MFIs (mean±SEM). HC, healthy controls; MFI, median fluorescence intensity; phospho-H2AX, phosphorylated H2AX; SLE, systemic lupus erythematosus.
Discussion
SLE is characterised by increased levels of cellular apoptosis, inefficient clearance of apoptotic cells, and the production of autoantibodies, which may arise from increased exposure to nuclear self-antigens derived from apoptotic debris.17–20 This SLE-associated increase in apoptosis is thought to be contributed by defective DNA repair mechanisms. Indeed, rare variants associated with SLE were shown to impair protein function of RNase H2, an essential enzyme that removes misincorporated ribonucleotides during DNA replication. The ineffective DNA damage repair that ensued resulted in increased expression of interferon (IFN)-regulated genes and an enhanced type-I IFN response in fibroblasts from patients.21 Furthermore, defective repair of DSBs has been associated with EBV-transformed B-cell lines in paediatric SLE and PBMCs in lupus nephritis.7 ,8 More recently, a polymorphism in the DNA repair gene RAD51B has been associated with increased risk of SLE.2
In this study, we explored DSB accumulation for the first time in SLE CD4+ T cells, CD8+ T cells and monocytes, compared with healthy controls and patients with RA. In addition, we measured intracellular DSB accumulation with detailed analyses of phospho-H2AX by flow cytometry, which can quantify DSBs with high specificity and sensitivity to genotoxicity (91% and 89%, respectively), while offering statistical superiority to other phospho-H2AX assays by analysing greater numbers of cells.22 ,23 The anti-phospho-H2AX antibody used in this study specifically detects phosphorylation of H2AX at serine 139, and has been previously validated and repeatedly used to accurately assess H2AX phosphorylation.21 ,24 ,25
For each of the analysed cell types, we revealed significantly increased DSB accumulation in patients with SLE compared with healthy controls. Further, DSB accumulation was significantly higher in SLE compared with RA, an inflammatory disease control. It remains to be seen, however, how this increase in DSBs in SLE compares with other conditions known to be associated with increased oxidative stress, such as sepsis and radiation therapy. As phospho-H2AX levels are known to be decreased in G1 compared with both S and G2 phases,26 ,27 we also assessed DSB accumulation at G0/G1, S and G2 phases to show significant DSB accumulation at each cell-cycle phase in SLE T cells and at G0/G1 and G2 phases in SLE monocytes. In combination, these findings suggest that increased DSB accumulation in SLE immune cells is independent of lineage and cell cycle.
Our analysis of the relationship between disease activity and DSB levels in patients with SLE showed a significant positive correlation in CD4+ T cells and CD8+ T cells, independent of the cell-cycle phases. When the SLEDAI scores were calculated without criteria for low complement binding and increased anti-dsDNA antibodies, the correlation was also significant in CD4+ T cells, CD8+ T cells and monocytes, independent of cell cycle. These data might support a proof of concept for the potential use of phospho-H2AX as a biomarker of disease activity in SLE. Although the utility of this biomarker will require replication efforts with a larger cohort for validation, the benefits of using phospho-H2AX as a biomarker for early detection, prognosis and treatment efficacy have been already described in cancer.28 ,29
Previous studies have shown that SLE lymphocytes and neutrophils are sensitive to oxidative stress.30 ,31 In this study, we assessed the DNA damage response to oxidative stress in specific immune cell types using a time-course treatment of H2O2. In CD4+ and CD8+ T cells from healthy individuals, our data showed that phospho-H2AX levels did not increase further with H2O2 exposures longer than 10 min. In addition, phospho-H2AX levels of healthy monocytes did not increase appreciably at any of the evaluated exposure times, indicating that monocytes may have an alternate or enhanced ability to handle oxidative stress. However, SLE phospho-H2AX levels continued to increase with H2O2 exposure times for each of the three immune cell subsets. These data suggest that SLE is associated with defective DSB repair or an intrinsically higher susceptibility for DNA damage in CD4+ T cells, CD8+ T cells and monocytes.
Taken together, these data provide evidence for defective repair of endogenous and oxidative stress-induced DSBs in SLE CD4+ T cells, CD8+ T cells and monocytes. In addition, DSB levels correlate with disease activity, and suggest further investigation of phospho-H2AX as a disease biomarker linking environmental exposures to the chromatin microenvironment in SLE. Importantly, our findings add to our current understanding of SLE, and offer further evidence for the role of aberrant DSB repair in disease pathogenesis.
Acknowledgments
The authors are grateful to the University of Michigan Lupus Program and the Michigan Lupus Cohort staff for their help with this study.
References
Footnotes
RN and PR contributed equally.
Contributors All authors listed contributed and fulfil authorship criteria as below: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work is appropriately investigated and resolved.
Funding The research included in this report was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases grant number T32AR007080-37 and the National Institute of Allergy and Infectious Diseases grant numbers R01AI097134 and U19AI110502.
Competing interests None declared.
Ethics approval University of Michigan IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.