Article Text

Abstract
Objective Previous studies have shown that differential DNA methylation is associated with SLE susceptibility. How DNA methylation affects the clinical heterogeneity of SLE has not been fully defined. We conducted this study to identify differentially methylated CpG sites associated with nephritis among women with SLE.
Methods The methylation status of 428 229 CpG sites across the genome was characterised for peripheral blood cells from 322 women of European descent with SLE, 80 of whom had lupus nephritis, using the Illumina HumanMethylation450 BeadChip. Multivariable linear regression adjusting for population substructure and leucocyte cell proportions was used to identify differentially methylated sites associated with lupus nephritis. The influence of genetic variation on methylation status was investigated using data from a genome-wide association study of SLE. Pathway analyses were used to identify biological processes associated with lupus nephritis.
Results We identified differential methylation of 19 sites in 18 genomic regions that was associated with nephritis among patients with SLE (false discovery rate q<0.05). Associations for four sites in HIF3A, IFI44 and PRR4 were replicated when examining methylation data derived from CD4+ T cells collected from an independent set of patients with SLE. These associations were not driven by genetic variation within or around the genomic regions. In addition, genes associated with lupus nephritis in a prior genome-wide association study were not differentially methylated in this epigenome-wide study. Pathway analysis indicated that biological processes involving type 1 interferon responses and the development of the immune system were associated with nephritis in patients with SLE.
Conclusions Differential methylation of genes regulating the response to tissue hypoxia and interferon-mediated signalling is associated with lupus nephritis among women with SLE. These findings have not been identified in genetic studies of lupus nephritis, suggesting that epigenome-wide association studies can help identify the genomic differences that underlie the clinical heterogeneity of SLE.
- Systemic Lupus Erythematosus
- Lupus Nephritis
- DNA methylation
- Epigenetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Lupus nephritis continues to cause substantial morbidity and increased mortality for individuals with SLE.1 The pathogenesis of lupus nephritis involves intrarenal factors such as immune complex formation and deposition that lead to complement activation, as well as extra-renal factors including genetic variants that predispose individuals to a loss of tolerance, leading to autoantibody production against nuclear antigens.2 In a genome-wide association study of lupus nephritis, the strongest genetic associations with lupus nephritis were observed in the PDGFRA-GSX2, SLC5A11 and ID4 regions. These results indicated that genetic variants outside of the major histocompatibility complex and not previously associated with SLE may have a greater influence in the development of nephritis among patients with SLE.3
More recently, DNA methylation, an epigenetic modification that influences gene expression, has been implicated in the pathogenesis of SLE. Two studies have indicated that hypomethylation of interferon-regulated genes in specific leucocyte subsets is associated with SLE susceptibility.4 ,5 Our own work has shown that among individuals with SLE, hypomethylation of interferon-regulated genes is associated with SLE-related autoantibody production.6 More recently, Coit et al7 also showed that, when compared with healthy controls, CD4+ T cells from patients with SLE having nephritis showed a greater degree of hypomethylation of interferon-regulated genes than patients with SLE not having nephritis.
In this study, we build on our previous work by conducting the largest genome-wide DNA methylation study of lupus nephritis to date to identify genes and pathways that are differentially methylated between SLE cases with and without nephritis. We also examine the influence of genetic variation on the identified associations, as well as methylation status of previously identified lupus nephritis susceptibility genes among patients with SLE with and without nephritis.
Methods
Study subjects
The primary subject group for this study comprised 80 patients with SLE having nephritis and 245 patients with SLE not having nephritis. All SLE cases were participants of the University of California, San Francisco Lupus Genetics Project and fulfilled at least four American College of Rheumatology classification criteria for SLE. To minimise confounding, all SLE cases were women of European descent who had never smoked. Lupus nephritis status was determined by medical record review. SLE cases were considered to have lupus nephritis if they fulfilled the renal classification criterion for SLE (persistent proteinuria >0.5 gm/day or >3+ on urine dipstick or presence of cellular casts)8 or had a renal biopsy showing lupus nephritis. A SLE case was considered not to have lupus nephritis if no evidence of renal involvement (eg, lacking haematuria and proteinuria) was identified on medical record review.
To replicate significant findings, we used the dataset generated by Coit et al.7 This study assessed the methylation status of CD4+ T cells from 56 women with SLE. A total of 28 women had lupus nephritis and 28 did not have lupus nephritis. The two groups were ethnically matched (17 European-American and 11 African-American individuals in each group). Additional details regarding this study group are provided in Coit et al.7 To assess for overlap between the two cohorts, we compared dates of birth and recruitment site for the individuals in each cohort, since genome-wide genetic data were not available for individuals in the replication cohort. Only one birth date was identical between the primary and replication cohorts and the individuals with this date of birth were recruited from different states within the USA. Thus, there is likely no overlap between the two cohorts.
DNA methylation assessment
DNA methylation profiling of genomic DNA from peripheral blood cells was performed using the Illumina HumanMethylation450 BeadChip, as previously described.6 This high-throughput platform assesses the methylation status for ∼480 000 CpG sites in ∼23 000 genes and assesses methylation for sites in promoters, 5′ and 3′ regions, gene bodies, CpG islands, CpG island shores and outside of CpG islands. For each site, the primary output is the β-value, which is the ratio of the methylated probe intensity to the overall probe intensity and represents the proportion of DNA that is methylated at that site (0=completely unmethylated, 1=completely methylated).
Prior to analysis, we performed extensive normalisation and quality control measures. We removed sites which had a low detection rate (p value for detection being <0.05) in more than 20% of samples (289 CpG sites). All samples met the detection threshold (<5% missing). To normalise the data, background signal was subtracted using the ‘noob’ method with the R package Methylumi.9 All sample mean normalisation was used to normalise colour channels.10 The final normalisation step was β-mixture quantile normalisation to normalise for differences between type 1 and type 2 probes on the Illumina Infinium platform.11 We also removed cross-reactive probes that may hybridise to more than one genomic location (30969), polymorphic probes which are known to contain a single nucleotide polymorphism (SNP) commonly found in populations of European ancestry (29 027) and non-CpG rs SNP probes (65).12 We calculated correlations between replicate samples (38 duplicate pairs) as a quality control measure and found r2>0.98 for all sites across the duplicate pairs. From the original dataset of 325 samples, we removed three samples that were identified as outliers through principal components analysis of methylation data. The final dataset consisted of 428 229 CpG sites assessed in 322 individuals with SLE, of whom 80 SLE cases had nephritis and 242 SLE cases did not have evidence of nephritis.
Methylation status of the 56 individuals used as the replication cohort was also assessed using the Illumina HumanMethylation450 BeadChip. Details regarding the data processing for this group are provided in Coit et al.7
Cell population estimation
DNA methylation patterns can vary between different blood cell populations.13 In order to account for cell-type heterogeneity between samples, we calculated principal components using the ReFACTor method14 to be used in multivariable regression. ReFACTor is a reference-free method that performs a principal component analysis (PCA) on the subset of CpG sites that are differentially methylated across different cell types to adjust for variance due to cell-type heterogeneity. We included the first two ReFACTor principal components in regression analyses to adjust for cell heterogeneity based on the examination of quantile-quantile plots for inflation and deflation of p values and calculation of genomic inflation factors (λ=0.90 for the first two principal components).
Population substructure
Population substructure may confound the associations between lupus nephritis and methylation since genetic ancestry may be associated with both methylation15 and disease status. Therefore, a genetic PCA was computed in EIGENSTRAT, as previously described,6 using SNP data generated with the Illumina HumanHap500 and ImmunoChip genotyping platforms. The first principal component was included in multivariable linear regression analysis to adjust for confounding due to population substructure.
Statistical analyses
All regression analyses were conducted in R (V.3.2.2) (R Core Team. R: A Language and Environment for Statistical Computing [program], Vienna, Austria, 2015. http://www.R-project.org/). For each of the 428 229 CpG sites under study, multivariable linear regression was used to determine the association between lupus nephritis status and methylation. For these regression models, methylation β-values were used as the outcome and lupus nephritis status was the primary predictor. Disease duration, the first genetic principal component and the first two ReFACTor principal components were included as covariates. The Benjamini-Hochberg false discovery rate (FDR) method16 was used to adjust for multiple comparisons, with an FDR q-value of <0.05 considered statistically significant.
All sites with an FDR q<0.05 (n=19) were further examined in an independent dataset generated by Coit et al7 for replication. For this dataset, association with lupus nephritis was determined using Wilcoxon rank-sum tests (STATA V.13.1, College Station, Texas, USA), with a stringent Bonferroni-corrected threshold of p<0.0026 (0.05/19) considered statistically significant.
To assess the role of genetic variation on the identified associations between DNA methylation sites and lupus nephritis, we used SNP data available from 264 of the study subjects from a genome-wide association study of SLE.17 We identified SNPs on the Illumina HumanHap500 BeadChip located 500 kb upstream and downstream of the associated CpG sites and analysed SNPs with a minor allele frequency of >0.05. For each CpG, we ran multivariable regression analyses as previously described and included the SNPs in the vicinity of the CpG as a covariate, assuming an additive model. Each CpG-SNP pair was analysed separately. FDR q-values were calculated based on the number of CpG-SNP pairs for a CpG site, with FDR q<0.05 considered statistically significant.
We further examined if methylation of previously identified lupus nephritis susceptibility genes3 was associated with nephritis among SLE cases. We examined the CpG sites on the HumanMethylation450 BeadChip that were within 5000 bp upstream and downstream of the genes. We readjusted the p values using FDR correction for this smaller subset of CpG sites from the final model in order to determine if any of these sites were differentially methylated between the two nephritis subgroups.
Last, we conducted a pathway analysis using the gometh() function in missMethyl R package to determine if differential methylation of genes in particular pathways were associated with lupus nephritis. This analysis used 1000 CpGs with the strongest evidence of association with lupus nephritis based on p values from multivariable regression models (without SNPs). The missMethyl package was developed specifically for the Illumina HumanMethylation450 array and the gometh() function tests for Gene Ontology enrichment for significant CpGs, taking into account the different number of methylation probes per gene on the array.
Results
The clinical characteristics of the 322 SLE cases examined in this study are presented in table 1. To minimise confounding by sex, race/ethnicity and smoking status, all study participants in the primary dataset were women of European descent who never smoked. SLE cases with nephritis had a longer disease duration and, not unexpectedly, a higher frequency of autoantibody positivity. Of the 50 SLE cases with nephritis who underwent renal biopsy and had pathology reports available for review, 32 (64%) had class III/IV (proliferative) nephritis and 10 (20%) had class V (membranous) nephritis.18
Characteristics of the 322 patients with SLE in this study
We first performed a genome-wide assessment of differential DNA methylation associated with lupus nephritis. Results from the multivariable regression analysis are shown in figure 1. The methylation status of 19 CpG sites had statistically significant evidence of association with lupus nephritis (FDR q<0.05) after adjusting for disease duration, population substructure and peripheral blood cell heterogeneity (table 2). Eight sites were more methylated in patients with SLE having nephritis compared with those without nephritis, with the difference in methylation ranging from 1.4% to 6.4%. Among these sites, the most hypermethylated site was cg23510764 on chromosome 5. Eleven sites were less methylated in SLE cases with nephritis compared with those without nephritis, with the difference in methylation ranging from −2.3% to −12.4% between SLE cases with and without nephritis. The most hypomethylated site among these 11 sites was cg05552874 located in IFIT1.
Differentially methylated CpG sites associated with nephritis in women with SLE

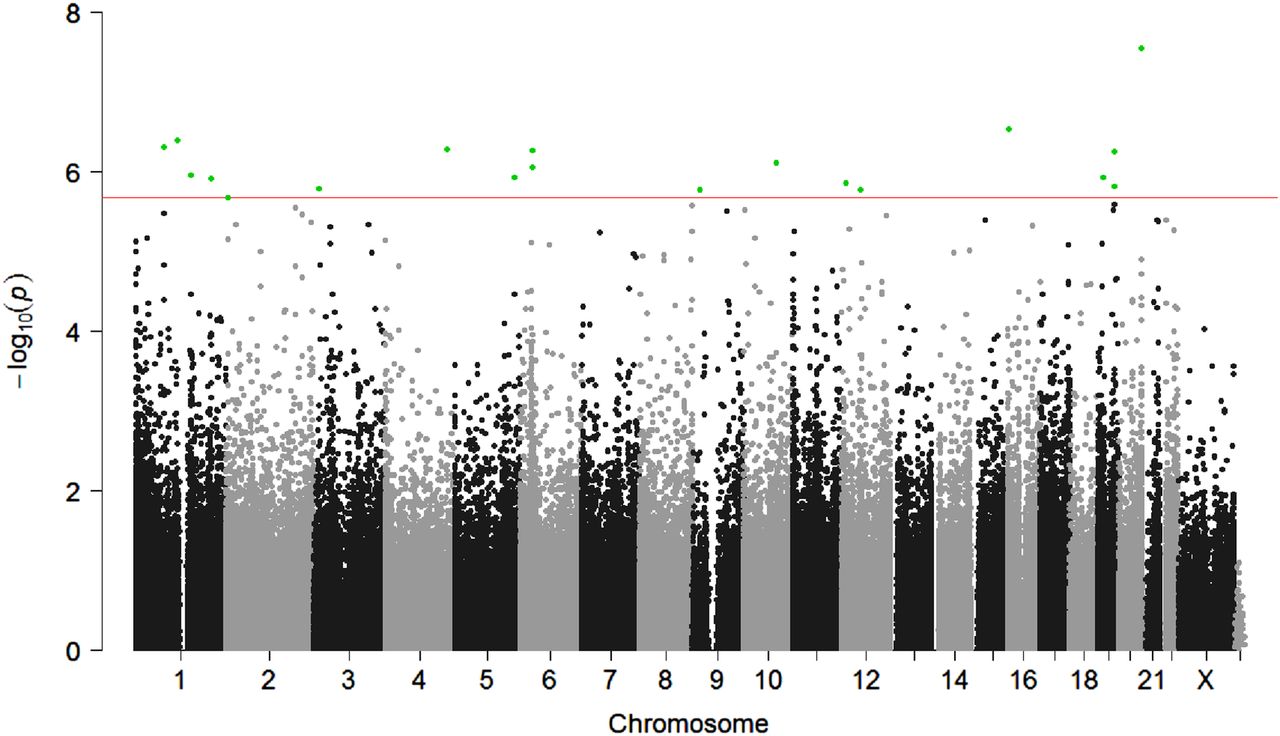{kind=link}
Manhattan plot of DNA methylation sites associated with lupus nephritis based on a multivariable linear regression. The red line shows a false discovery rate (FDR) cut-off of significance level p<2.1E-06. Significant CpG sites are highlighted in green.
We then sought replication for these findings using DNA methylation data generated with the same platform from CD4+ T cells from 56 patients with SLE.7 Out of the 19 sites we identified in our initial analyses, methylation of four sites in or near hypoxia-inducible transcription factor (HIF)3A, interferon-induced protein 44 (IFI44) and lacrimal proline-rich protein 4 (PRR4) in CD4+ T cells showed evidence of association with lupus nephritis in Wilcoxon rank-sum tests (p<0.0026), as shown in table 2.
Next, we examined whether methylation of 15 previously identified lupus nephritis susceptibility genes3 was associated with lupus nephritis, to determine if epigenetic associations are driven by genetic variation. We identified 12 794 CpG sites that were located within 5000 bp upstream or downstream of the lupus nephritis susceptibility genes for this analysis. Even with readjustment of the FDR q-value for only these sites, no site showed evidence of association with lupus nephritis (FDR q>0.1, data not shown).
To determine if the associations between methylation status and lupus nephritis could be confounded by genetic variation (ie, that genetic variation influences both the development of lupus nephritis and extent of methylation at a site), we used genome-wide SNP data available on 264 study subjects who had been previously genotyped with the Illumina HumanHap500 BeadChip. SNPs within 500 kb of the associated CpG site were included in multivariable regression analyses. Online supplementary table S1 shows the total number of SNPs studied for each CpG site and the number of SNPs with FDR q<0.05 in multivariable regression modelling. SNPs in or near HELZ2, HIF3A, MIR-29B-2/LOC148696 and PRR4 showed evidence of association with methylation (FDR q<0.05) but did not substantially alter the association between methylation and lupus nephritis status (see online supplementary table S2). These findings suggest that the observed associations between methylation status and lupus nephritis do not appear to be driven by genetic variation of these genes.
supplementary tables
We also examined whether these results could be confounded by medication exposure. While we were unable to adjust for medication dose at sample collection (data not available), we ascertained previous exposure to prednisone, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil and cyclophosphamide by medical record review and patient questionnaire. While medication exposure did differ between those with and without lupus nephritis as expected (eg, higher prevalence of mycophenolate mofetil and cyclophosphamide use in patients with SLE having lupus nephritis compared with patients with SLE not having nephritis), adjusting for previous medication exposure did not significantly alter the associations between methylation of 19 sites in table 2 and lupus nephritis (data not shown).
Last, we performed a pathway analysis to determine if genes with evidence of differential methylation associations with lupus nephritis represent particular biological pathways. Using Gene Ontology as the basis for this analysis, the top 1000 CpG sites with the strongest evidence of association by regression p value were studied. Thirty-three Gene Ontology groups were found to be over-represented in the top 1000 CpG sites. The 10 most significantly associated Gene Ontology groups as determined by FDR q-value are shown in table 3. These results indicate that differential methylation of genes involved in interferon signalling and host response pathways show the strongest evidence of association with lupus nephritis among individuals with SLE.
The top 10 Gene Ontology groups represented by the 1000 CpG sites with the strongest evidence of association with lupus nephritis
Discussion
In this study, we present the largest genome-wide assessment of DNA methylation in lupus nephritis to date. In our initial samples obtained from whole blood, we identified evidence for differential methylation of 19 sites in 18 genomic regions associated with lupus nephritis. Association with four sites in HIF3A, IFI44 and PRR4 were subsequently replicated when we examined DNA methylation of CD4+ T cells from an independent set of women with SLE. Observed methylation associations are not driven by DNA sequence variation surrounding the associated CpG site. In pathway analyses, differential methylation of interferon signalling and host defence genes had the strongest evidence of association with lupus nephritis.
Two of the replicated associations, cg22891070 and cg16672562, are located approximately 200 bp upstream from the transcription start site or within the gene body of HIF3A, depending on the gene transcript. HIF3A encodes the third isoform of the α subunit of the HIF, which can activate the transcription of over 100 genes involved with the cell's response to hypoxia, including genes involved in angiogenesis, glucose metabolism and cell proliferation. The role of HIF3A is less well delineated compared with the two other isoforms, in part because HIF3A mRNA undergoes alternative splicing, with at least seven known variants. Specific variants of HIF3A have been shown to act as a negative regulator of HIF1 and HIF2, while overexpression of other variants upregulate specific HIF target genes.19 Hypermethylation (instead of the hypomethylation observed in this study) of these CpG sites has been associated with increased body mass index.20 Previous epidemiological studies have suggested that increased body mass index is associated with chronic kidney disease,21 even among patients with SLE.22 Thus, it is not evident that the associations between lupus nephritis and hypomethylation of HIF3A is due to confounding by body mass index. Chronic hypoxia in the tubulointerstitium of the kidney is proposed to be a common pathway that leads to end-stage renal disease and thus HIF plays a critical pathophysiological role in this process. HIF is also being studied as a target for therapies to prevent progression of chronic kidney disease.23 Our findings for HIF3A are novel, since neither genetic variation nor methylation of HIF genes has been associated with SLE or specific SLE-related sub-phenotypes in prior studies. Hypomethylation of HIF-related genes may contribute to the development of lupus nephritis or occur as the result of organ damage from nephritis in patients with SLE. In either scenario, therapy targeting the HIF pathway may provide a novel strategy to decrease the progression of kidney damage for patients with lupus nephritis.
We also replicated an association between lupus nephritis and hypomethylation of cg01079652, which is located within the gene body of IFI44. While the role of IFI44 is not completely defined, some evidence suggests that this interferon-inducible gene acts as a suppressor of viral replication.24 In the study by Coit et al,7 hypomethylation of this CpG site was identified when comparing SLE cases with nephritis with healthy controls and was not identified when SLE cases without nephritis were compared with healthy controls. In a study by Absher et al,5 this site was significantly hypomethylated in CD4+ T cells, CD19+ B cells and CD14+ monocytes when comparing patients with SLE with healthy controls. Of note, hypomethylation of IFI44L, a paralog of IFI44, was found to be associated with SLE,4 SLE-related autoantibody production6 by our group and more recently was shown to be a biomarker for SLE when compared with individuals with other autoimmune diseases and healthy controls.25
The third replicated association was with cg23256579, which occurs ∼1500 bases upstream or within the body of PRR4, depending on the transcript variant. PRR4 has been identified as a potential biomarker of the dry-eye syndrome.26 Read-through transcription also exists between this gene and the upstream gene PRH1 (proline-rich protein HaeIII subfamily 1). This transcript is a candidate for nonsense-mediated mRNA decay and is unlikely to produce a protein product. No previous associations between methylation of this site and SLE-related phenotypes have been reported.
We also found evidence of differential methylation of 15 CpG sites near or within HELZ2, COR07, CASQ2, DDX60, FKBP5, IFIT1, IL6R, MIR-29B-2b, SLC6A6, METTL7A, MLLT3 and RSAD2 in whole blood, which did not meet our stringent threshold for replication in the CD4+ T-cell dataset. Of the 15 sites, 13 showed concordant direction of differential methylation between the initial and replication cohorts. Two sites, located in/near in IFIT1 and FKBP5 had suggestive statistical evidence of association in the replication cohort (p=0.0069 and p=0.024, respectively). While a lack of replication may indicate false-positive results, these findings may not have replicated due to insufficient statistical power given the small sample size of the replication dataset, the differences in race/ethnic compositions between the two sample groups or because the differential methylation observed for these sites are not driven by CD4+ T cells. Differential methylation of these genes in other cell populations may be more relevant for the development of lupus nephritis. Differential methylation of sites near or within IFIT1 and RSAD2 have been previously associated with SLE-related autoantibody production,6 while differential methylation of the associated sites in HELZ2 (also known as PRIC285), DDX60, IFIT1, CASQ2, IFI44 and FKBP5 has been observed in specific leucocyte subsets among patients with SLE compared with healthy controls.4 ,5 Thus, while the p value for association with lupus nephritis for these sites did not meet our threshold for replication, methylation of many of these sites is likely associated with SLE-related outcomes given their previously identified association with SLE susceptibility.
Previously, we showed that methylation of CpG sites in 11 genes was associated with anti-double-stranded (ds)DNA and SLE-related autoantibody production and 7 out of the 11 genes either induced or regulated type 1 interferon signalling. Given the strong correlation between anti-dsDNA autoantibodies and lupus nephritis, we expected to see interferon-related genes dominate the list of statistically significant results. However, among the 18 genomic regions with the strongest statistical evidence of association with lupus nephritis in this study, only four are known to be induced by type 1 interferon (IFI44, IFIT1, DDX60 and RSAD2). Thus, in contrast to SLE-related autoantibodies, the majority of the most significantly associated methylation sites with lupus nephritis are not in interferon-related genes. This finding may represent a key pathophysiological difference between autoantibody production and lupus nephritis. However, interferon-related genes do likely influence the development of lupus nephritis to some extent, given the results of the pathway analyses.
Associations with DNA methylation and lupus nephritis were also examined by Coit et al.7 However, their results are not directly comparable with ours given differences in study design—instead of performing a within-SLE case analysis as in this study, SLE cases with and without nephritis were compared with healthy controls. However, similar to our findings, Coit et al observed that interferon-related genes were even less methylated in SLE cases with lupus nephritis than SLE cases without nephritis when both groups were compared with healthy controls.
Strengths of our study include the large sample size and selection of SLE cases to minimise confounding due to sex, ethnicity and smoking status. Unlike many studies of DNA methylation, we were able to adjust for differences in genetic ancestry among the SLE cases using genome-wide SNP data. Our analyses also accounted for disease duration to mitigate the effect of the disease itself on DNA methylation status and the potential misclassification of lupus nephritis status. Last, we employed a reference-free method to estimate and adjust for potential differences in the leucocyte composition between study subjects.
Limitations of our study include its cross-sectional nature. Thus, we are unable to determine if these changes in methylation occur prior to or after the development of lupus nephritis. However, the identification of novel disease-relevant pathways provides insights into the pathogenesis and progression of lupus nephritis, even if these pathways are not the biological processes that initiate disease. In addition, the body mass index of the participants was not available to include in our analyses. Last, we examined DNA from peripheral blood cells since it can be easily collected in a clinical setting for the development of future clinical tests and is readily available from our study collection. Therefore, we could not assess which specific leucocyte population harbours these DNA methylation changes. However, our findings for CpG sites within or near HIF3A, PRR4 and IFI44 were replicated in a dataset derived from CD4+ T cells, which suggests that methylation changes in CD4+ T cells contribute to the differences we identified in our study.
In summary, we have shown that hypomethylation of CpG sites in or near tissue hypoxia and interferon signalling response genes is associated with lupus nephritis. Our results support the premise that genetic and epigenetic variations can be independently associated with disease manifestations. Thus, epigenome-wide association studies can identify novel pathways associated with disease manifestations not identified by genome-wide association studies and can provide complementary insight into the pathogenesis and progression of genetically complex autoimmune diseases.
References
Footnotes
Contributors Conceived and designed the experiments: SAC, LFB and LAC. Performed the experiments: HLQ, PC, AHS and LFB. Analysed the data: AM, OS, RRN, JN and SAC. Contributed reagents/materials/analysis tools: PC, HLQ, AHS, LFB and LAC. Wrote the paper: AM, OS and SAC.
Funding The work was supported by the following NIH (http://www.nih.gov) grants: R01 AR052300 (LAC), K24 AR02175 (LAC), P60 AR053308 (LAC), R01 AI097134 (AHS), UCSF-CTSI KL2 RR024130 (SAC), UCSF-CTSI UL1 TR000004 (SAC) and K23 AR063126 (SAC); Alliance for Lupus Research (http://www.lupusresearch.org, LAC); Mary Kirkland Scholar Award (http://www.hss.edu/mary-kirkland-scholar-program.asp, LAC); Rheumatology Research Foundation (AM), the Irene Perstein Award (https://medschool.ucsf.edu/school-medicine-irene-perstein-award/, SAC), and the Russell/Engleman Rheumatology Research Center (https://russellenglemancenter.ucsf.edu/, RRN, JN, LAC and SAC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Competing interests None declared.
Ethics approval Institutional Review Board, University of California, San Francisco, USA.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Methylation data for the study participants are available at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000947.v1.p1. Additional phenotype data is available to academic investigators by contacting SAC, the corresponding author.