Article Text

Abstract
Objectives To examine microparticles (MPs) from patients with SLE and healthy controls (HCs) by determining the cellular origin of the MPs, quantifying attached fragments of complement component 3 (C3) and assessing the ability of MPs to bind to circulating phagocytes and erythrocytes. These features may be relevant for clearance of MPs in SLE pathogenesis.
Methods Attached C3 fragments (C3b, iC3b, C3d), membrane integrity and cell surface markers of MPs from 18 patients with SLE and 11 HCs were measured by adding specific antibodies, 7-aminoactinomycin D (7AAD) and annexin V. MPs from all subjects were labelled with carboxyfluorescein diacetate succinimidyl ester and allowed to bind to autologous phagocytes and erythrocytes in the presence of autologous serum, and the binding to individual cell populations was assessed by flow cytometry.
Results The proportion of MPs bearing C3 fragments was higher in patients with SLE than in HCs (p=0.026), but the amount of opsonising C3b/iC3b molecules was lower (p=0.004). The C3b/iC3b level correlated with the concentration of circulating C3 (rs=0.53, p=0.036). Phagocytes and erythrocytes from patients and HCs bound autologous MPs, and granulocytes from patients bound 13% more MPs than those from HCs (p=0.043). The presence of erythrocytes inhibited the MP binding to granulocytes by approximately 50%.
Conclusions Our demonstration of altered composition of C3 fragments on MPs from patients with SLE, including decreased numbers of opsonising C3 fragments, and competitive binding of MPs to circulating phagocytes and erythrocytes corroborates the hypothesis of defective clearance of apoptotic material in SLE, and indicates that differences in both MP opsonisation and binding of MPs to cells are important in the pathogenesis of SLE.
- Autoimmunity
- Apoptosis
- Complement
- Microparticles
- Systemic Lupus Erythematosus
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Microparticles (MPs) are hypothesised as central in the pathogenesis of SLE, but the mechanisms involved are unclear. MPs are small membrane-bound vesicles (0.1–1 µm) released from activated or apoptotic cells (ACs).1 They carry, among other, nuclear antigens triggering the production of various antinuclear antibodies, a hallmark of SLE.
SLE is a systemic autoimmune disease characterised by activation of the innate immune system and activation of T and B lymphocytes, leading to production of autoantibodies, immune complex (IC) deposits and complement-mediated tissue damage.2 This results in clinical manifestations from various tissues, of which malar rash, non-erosive arthritis and nephritis are among the most common.3 The disease can range from mild to severe with varying degrees of organ damage and is associated with an overall increased mortality.4
MPs have surface similarities with both ACs and ICs because the MP surface contains both phosphatidylserine, other cell surface markers and antigen–antibody complexes.5 Accordingly, because SLE is associated with deficient clearance of AC material6–8 and ICs,9 clearance of MPs is likely to be impaired in SLE. Clearance of ACs is normally a non-inflammatory process; ACs are rapidly phagocytised following recognition via receptors for opsonins, phosphatidylserine or β2-glycoprotein I, exposed on the cell membrane.7 ,10 ,11 ICs are cleared by binding to erythrocytes via complement receptor type 1 (CR1) that binds C3b, C4b and, with lesser affinity, iC3b on the IC.12 The ICs are subsequently transported to the fixed mononuclear phagocytic system in liver, spleen or bone marrow.6 ,9 ,12–15 Based on this, and previous findings of neutrophil MP binding to erythrocytes, we found it reasonable to suggest that MPs might be cleared in the same manners.16 Deficiencies in these clearance mechanisms are likely to be impaired in patients with SLE. Several hypotheses about AC clearance defects exist. One pertains to defects in the receptor-mediated recognition by phagocytes.7 ,17 Another focuses on defects in macrophage phagocytosis.8 Third, diminished clearance of ACs in SLE is linked to deficiencies in complement components or complement receptors. For example, one third of patients with SLE have antibodies against complement, and deficiencies of components of the classical pathway are strongly associated with SLE,6 especially C1q deficiency.18 Clearing of ICs is also compromised in patients with SLE, who express fewer CR1 molecules per erythrocyte than healthy controls (HCs). Deficient MP clearance might lead to accumulation of MPs with disrupted membrane integrity and exposure of antigenic cell material to the immune system, resulting in a proinflammatory response.7
The qualitative properties of MPs in patients with SLE are sparsely investigated. It has been demonstrated that in vitro generated MPs carry double-stranded DNA (dsDNA) on their surface, and that antibodies from SLE plasma bind to such MPs. In addition, the surface of MPs from patients with SLE contains large amounts of IgG, which correlate with the individual levels of circulating anti-dsDNA antibodies.5 ,19 ,20 This study aimed to increase the knowledge about mechanisms for clearance of MPs including their binding to circulating blood cells, and to determine differences between patients with SLE and HCs in this respect.
Materials and methods
Patients with SLE and HCs
The patients with SLE were included either as they were hospitalised with active disease or from our outpatient rheumatology clinic, and they fulfilled internationally accepted classification criteria.21 ,22 We aimed to include both patients with disease-active and disease-inactive SLE, to be able to compare these two groups. Patients who had a malignant disease, were pregnant or received cytostatics or high-dose prednisolone (>25 mg/day) were excluded. We used the scoring index SLE Disease Activity Index (SLEDAI) 2K 30 days for estimation of disease activity,23 and we defined active disease as SLEDAI score ≥5. The HCs examined were blood donors at the Blood Bank at Copenhagen University Hospital Rigshospitalet. The study was approved by the Regional Scientific Ethics Committee (Protocol no. H-1-2013-046).
Eighteen patients with SLE, of which 8 had active disease (SLEDAI ≥5) and 11 HCs, were included in the study. The patient group consisted of 83% women, compared with 82% in the HC group, and the age was median (range) 36 (22–69) and 34 (24–54) years, respectively.
Purification of MPs
We collected 9 mL of blood in a K3EDTA tube (Vacuette, Greiner Bio-One GmbH) by venous puncture. The blood was centrifuged at 1800×g for 10 min at 37°C for cell removal. The supernatant was moved to a Falcon tube and centrifuged at 3000×g for 10 min at 37°C for removal of most of the platelets. The remaining platelet poor plasma was filtered through a 1.2 µm syringe filter (Minisart, Sartorius) and divided into aliquots of 460 µL in Eppendorf tubes. Forty microlitres of Roswell Park Memorial Institute medium 1640 (RPMI) was added to each tube. After centrifugation at 19 000×g for 30 min at 21°C, 475 µL supernatant was removed, leaving 25 µL in the tube. Then 225 µL RPMI, filtered through a 0.1 µm filter (Minisart), was added, and the MPs were resuspended in a total volume of 250 µL. After a second centrifugation at 19 000×g for 30 min at 21°C, 225 µL was removed, leaving 25 µL in the tube. Seventy-five microlitres of the filtered RPMI was added, and the MPs were resuspended in a total volume of 100 µL per aliquot. We stored the purified MPs in an incubator at 37°C for 20–24 hours until analysis or incubation with blood cells.
Analysis of MPs
For determination of the cellular origin of MPs, two 50 µL aliquots of MPs were added to 40 µL of filtered RPMI and incubated with (1) 5 µL allophycocyanin (APC) conjugated anti-CD3 (T cells), 10 µL phycoerythrin (PE)-conjugated anti-CD61 (platelets), 5 µL fluorescein isothiocyanate (FITC)-conjugated anti-CD146 (endothelial cells); and (2) 3 µL peridinin chlorophyll protein complex (PerCP)-conjugated anti-CD14 (monocytes), 3 µL anti-CD15-APC (neutrophils), 5 µL anti-CD19-PE (B cells). For examination of membrane integrity and exposure of chromatin, one 50 µL aliquot of MPs were added to 5 µl annexin V and 3 µL 7-aminoactinomycin D (7AAD), respectively. All antibodies were from Becton Dickinson (BD), except anti-CD61-PE (Biolegend). All incubations took place for 30 min, except for 7AAD, which incubated for 5 min. The stained MPs were further diluted with 0.1 µm filtered phosphate-buffered saline (PBS) solutions: calcium-containing PBS for the tube with annexin V and citrate-containing PBS for the remaining tubes.17 For quantification, we used BD TruCount beads (BD), according to the manufacturer's instructions.
The purified MPs were evaluated by flow cytometry (BD FacsCalibur) with all detectors in logarithmic mode. CellQuest software (BD) was used for acquisition, and we applied Flow Jo software V.7.6.5 (Tree Star) for analysis. MPs were defined as particles with a diameter of 0.1–1 µm, and this range in forward scatter was determined by the aid of Fluoresbrite (Polysciences) size beads of 0.1 and 1 µm (figure 1). Unstained samples were used as controls.

Gating of microparticles (MPs). (A) Flouresbrite 0.1 µm beads added to PBS were analysed by flow cytometry to set a 0.1 µm lower limit. (B) Flouresbrite 1.0 µm beads added to PBS were analysed by flow cytometry to set an upper 1.0 µm limit. (A and B) The final gate for MP analysis is shown. (C and D) Example of gating of stained MPs, including the gate used for analysis, where panel C shows the ungated forward/sidescatter (FSC/SSC) with the MP gate, panel D shows the unstained negative control and panel E shows MPs stained with anti-CD61 PE.
Analysis of C3 fragments on the surface of MPs was done by means of antibodies against specific C3 fragments: MPs incubated with FITC-conjugated polyclonal rabbit anti-C3d antibodies (Dako) for 30 min for assessment of all surface-bound C3 fragments.24 Unstained samples were used as negative controls. Incubation with monoclonal antibodies (mAb) recognising C3b and iC3b (clone f1-23) or iC3b alone (clone f1-7) for 30 min, followed by staining with a PE-conjugated secondary antibody (Dako) for 15 min, was used for quantification of these C3 fragments.24 An irrelevant in-house generated non-reacting antibody in culture supernatant was used together with the PE-conjugated secondary antibody as negative control. All samples were analysed by flow cytometry.
Binding of MPs to blood cells
MPs from 18 patients with SLE and 10 HCs were loaded with the fluorescent substrate 5 (and 6-)-carboxyfluorescein diacetate succinimidyl ester (CFSE) and incubated with autologous cells: leucocytes, purified from 100 µL whole blood subjected to erythrolysis with Easy Lyse (Dako; a 4.5% solution), or 100 µL washed whole blood, or 1 µL diluted (1:100) whole blood cells. Autologous serum (30% v/v) was present, and all sera were frozen within 60 min after collection. After incubation for 1, 5, 15, 30 and 60 min, respectively, 10 µL of the solution was transferred to 1 mL PBS on ice. The binding of MPs to granulocytes, monocytes and erythrocytes was measured by flow cytometry as the CFSE median fluorescence intensity (MFI) of each cell population.
Within morphological gates based on forward and side light scatter characteristics, and by staining with anti-CD45−PerCP, anti-CD15−APC and anti-CD14−PE (all from BD), granulocytes were identified as CD45+ and CD15+ cells; monocytes were identified as CD45+ and CD14+ cells; and erythrocytes were identified as CD45−, CD14− and CD15− cells.
Statistical analysis
Differences between HCs, patients with disease-inactive SLE and patients with disease-active SLE were performed using the Kruskal-Wallis test, and the Mann-Whitney U test was used when patients with SLE and HCs were compared. Correlations were determined by Spearman's correlation coefficient (rs). The ability of MPs to bind to cells was assessed by a linear mixed model for longitudinal measurements, where the logarithm of the CFSE MFI was used as dependent variable, and the influence of incubation time and group was explored; binding was regarded significant if the influence of incubation time was statistically different from the zero value. The inhibition of binding exhibited by erythrocytes was calculated as the percentage of binding relative to binding without erythrocytes, and was shown as mean values with 95% CIs. If the interval did not overlap 100%, the inhibition was considered statistically significant. Statistical analyses were done by means of SAS Software v. 7.1 (SAS Institute), and figures were created in GraphPad Prism (GraphPad Software).
Results
Clinical characteristics of patients with SLE
The patients' median (range) SLEDAI score was 4 (0–18). Five of the patients with active disease (n=8) had biopsy-verified lupus nephritis, one had vasculitis and transient proteinuria, one had arthritis and pleurisy and one had arthritis as the only clinical symptom. One patient with inactive disease was known to have C1q deficiency.
Surface characteristics of MPs from patients with SLE and HCs
The total number of MPs per millilitre whole blood did not differ between patients with disease-active SLE, patients with disease-inactive SLE and HCs, and neither did the distribution of MPs concerning cell surface markers and membrane integrity: CD3 (p=0.36), CD14 (p=0.82), CD15 (p=0.38), CD19 (p=0.96), CD61 (p=0.44), CD146 (p=0.41), AnxV (p=0.71) and 7AAD (p=0.98) (figure 2, see online supplementary table for exact figures). As expected, platelet-derived CD61+ MPs were the most frequent MP type in patients with SLE and HCs.

Number of microparticles (MPs) and distribution of MP surface markers in patients with SLE and HCs. Three aliquots of MPs were incubated with (1) anti-CD3, anti-CD61 and anti-CD146; (2) anti-CD14, anti-CD15 and anti-CD19; and (3) 7AAD and annexin V, respectively. All incubations took place for 30 min, except for 7AAD, which incubated for 5 min. During subsequent flow cytometric analysis, MPs were defined as particles with a diameter of 0.1–1 µm, determined by size beads (Fluoresbrite). For calculation of numbers of MPs per millilitre full blood, BD TruCount beads were added. (A) The concentration of MPs per millilitre whole blood, (B) the number of MPs binding 7AAD or annexin V, and (C and D) the number of MPs positive for different surface markers are shown for patients with disease-active (triangles) and patients with disease-inactive (squares) SLE and for HCs (circles). Bars and error bars represent median and range.
supplementary methods
The percentage (range) of MPs bearing C3 fragments, as measured by anti-C3d, was higher in patients with SLE than in HCs (69.3% (52.1, 94.5) vs 65.0% (50.2, 74.6); p=0.026). In addition, the distribution of C3 fragments differed between patients with SLE and HCs; the median (range) MFI arising from binding of mAb f1-23, recognising both C3b and iC3b, to the surface of MPs from patients with SLE was lower than the corresponding MFI of MPs from HCs (202 (54, 1034) vs 477 (278, 1452); p=0.0044). The C3b+iC3b level correlated with the level of circulating C3 in the patients measured by routine methods (rs=0.53, p=0.036) (figure 3).
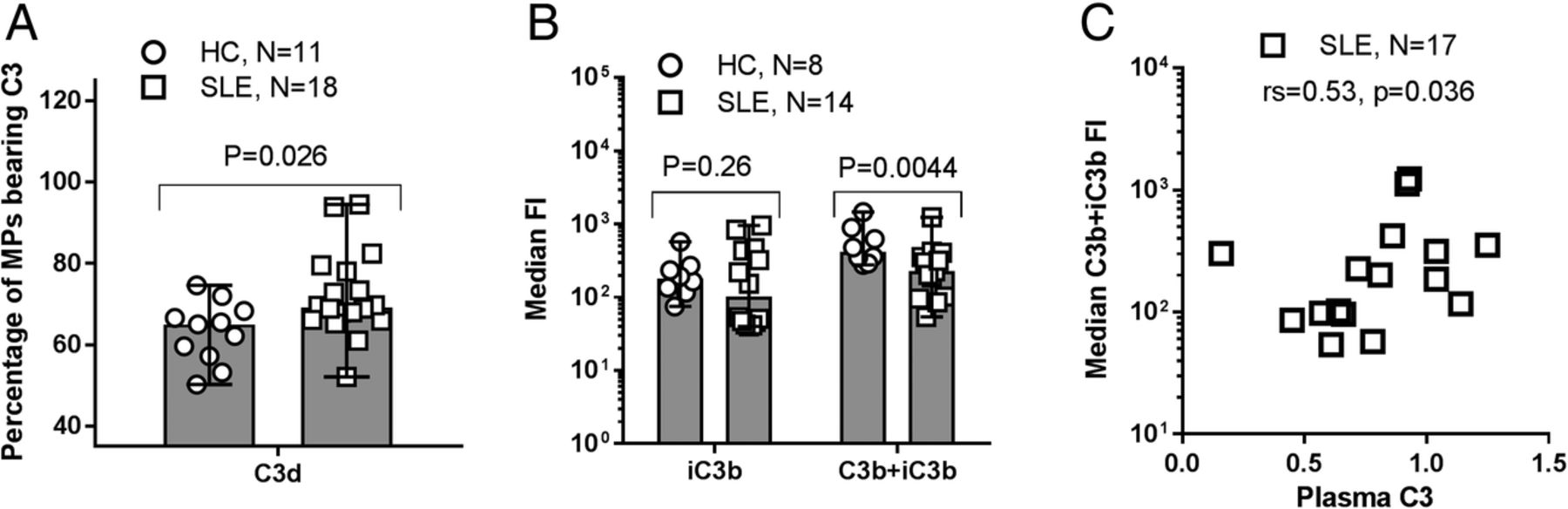
C3 fragment deposition on microparticles (MPs) from patients with SLE and healthy controls (HCs). (A) Purified MPs from patients with SLE and HCs were incubated with FITC-conjugated, polyclonal rabbit anti-C3d (recognising all C3 fragments) for 30 min and analysed by flow cytometry. Unstained MPs were used as negative controls to distinguish between C3 fragment-positive and negative MPs. The percentages of MPs bearing C3 fragments are shown as median (range). (B) Aliquots of purified MPs from HCs and patients with SLE were incubated for 30 min with monoclonal antibodies recognising C3b and iC3b (clone f1–23) and iC3b alone (clone f1–7), respectively. After staining with PE-conjugated secondary antibody, the samples were analysed by flow cytometry. A non-reacting control for the primary antibody was used to define C3 fragment-positive MPs, the fluorescence intensity (FI) of which is shown as median (range). Data from one experiment are missing due to total absence of events during flow cytometry. (A and B) MPs were defined as all particles with a diameter of 0.1–1 µm by the aid of size beads (Fluoresbrite). (C) The relationship between the circulating level of C3 and the median C3b+iC3b fragment-derived FI is shown for patients with SLE.
Binding of MPs to phagocytes and erythrocytes
Significant binding of MPs to granulocytes (p<0.0001), monocytes (p<0.0001) and erythrocytes (p<0.0001) was observed (figures 4 and 5). There were no statistically significant differences between patients with disease-active SLE, patients with disease-inactive SLE and HCs regarding binding of MPs to granulocytes (p=0.11), monocytes (p=0.84) or erythrocytes (p=0.73). However, when comparing the whole population of patients with SLE with HCs, a 13% higher binding of MPs to granulocytes was found in the patient group (p=0.043).

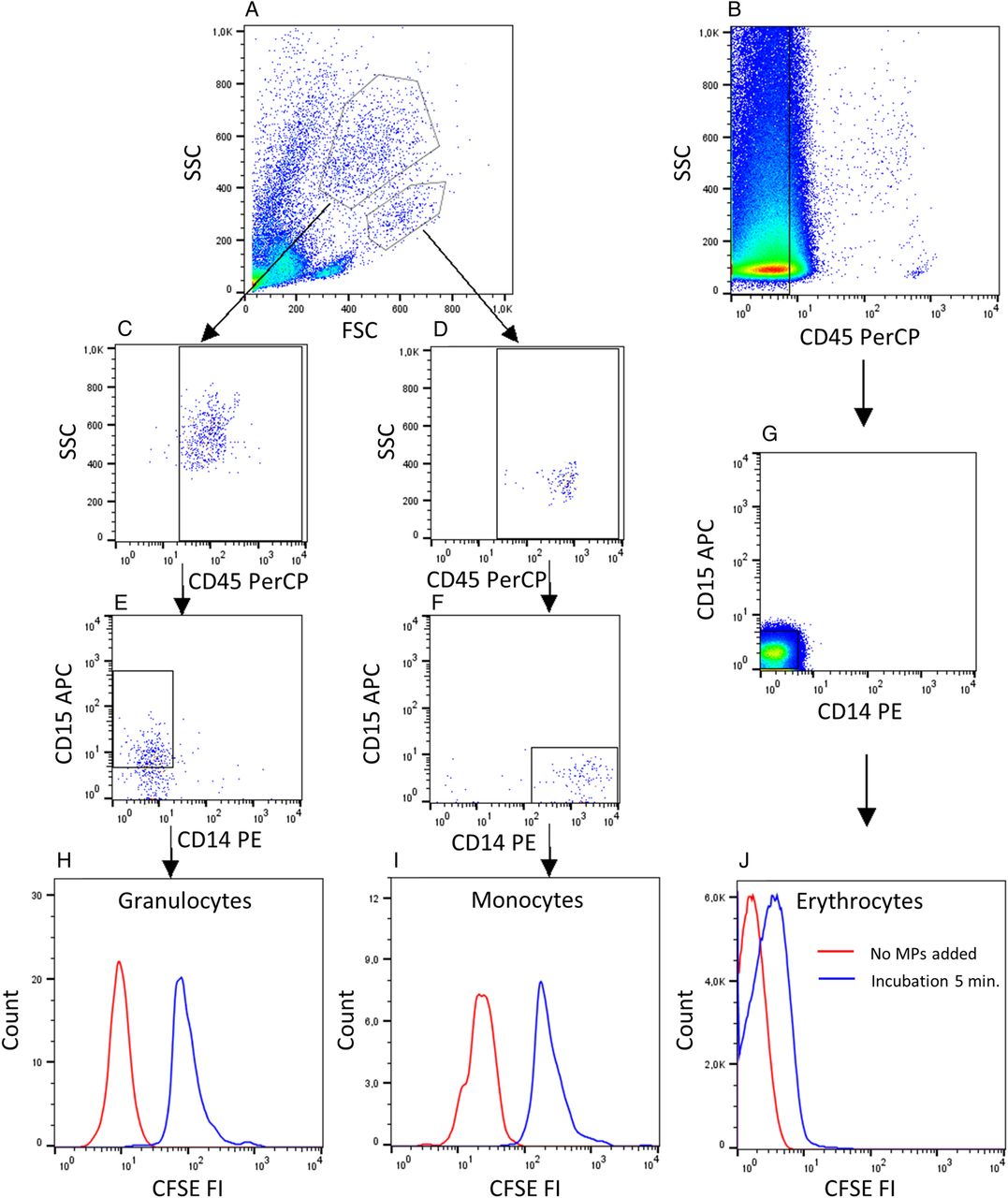
Binding of microparticles (MPs) to granulocytes, monocytes and erythrocytes. (A) Isolated leucocytes or (B) whole blood cells diluted (1:100) were incubated with autologous CFSE-labelled MPs for 5 min in the presence of 33% v/v autologous serum. (C and D) The gating strategy for granulocytes and monocytes included gating for CD45+ cells and (E and F) positivity for CD15 or CD14, respectively. (B and G) Erythrocytes were gated as cells negative for CD45, CD14 and CD15. (H–J) The background fluorescence intensity (FI) after no addition of MPs (red line) and FI after binding of MPs (blue line) are shown. Data are representative of experiments involving 10 HCs and 18 patients with SLE. CFSE, carboxyfluorescein diacetate succinimidyl ester; FSC, forward scatter.

Binding of microparticles (MPs) to phagocytes and erythrocytes in the presence of autologous serum. Preparations of (A and B) purified leucocytes, where the erythrocytes had been removed by lysis, and (C) full blood diluted 1:100 in RPMI from HCs (circles), patients with disease-active (triangles) or patients with disease-inactive (squares) SLE were incubated with carboxyfluorescein diacetate succinimidyl ester-labelled MPs for 60 min in the presence of 33% (v/v) autologous serum. The binding of MPs to (A) granulocytes, (B) monocytes and (C) erythrocytes is shown as the mean (95% CI) of the median fluorescence intensity (MFI) after subtraction of the background fluorescence observed when no MPs were added. One experiment involving a healthy donor has been excluded due to lack of binding to all cell types.
Ability of erythrocytes to inhibit binding of MPs to phagocytes
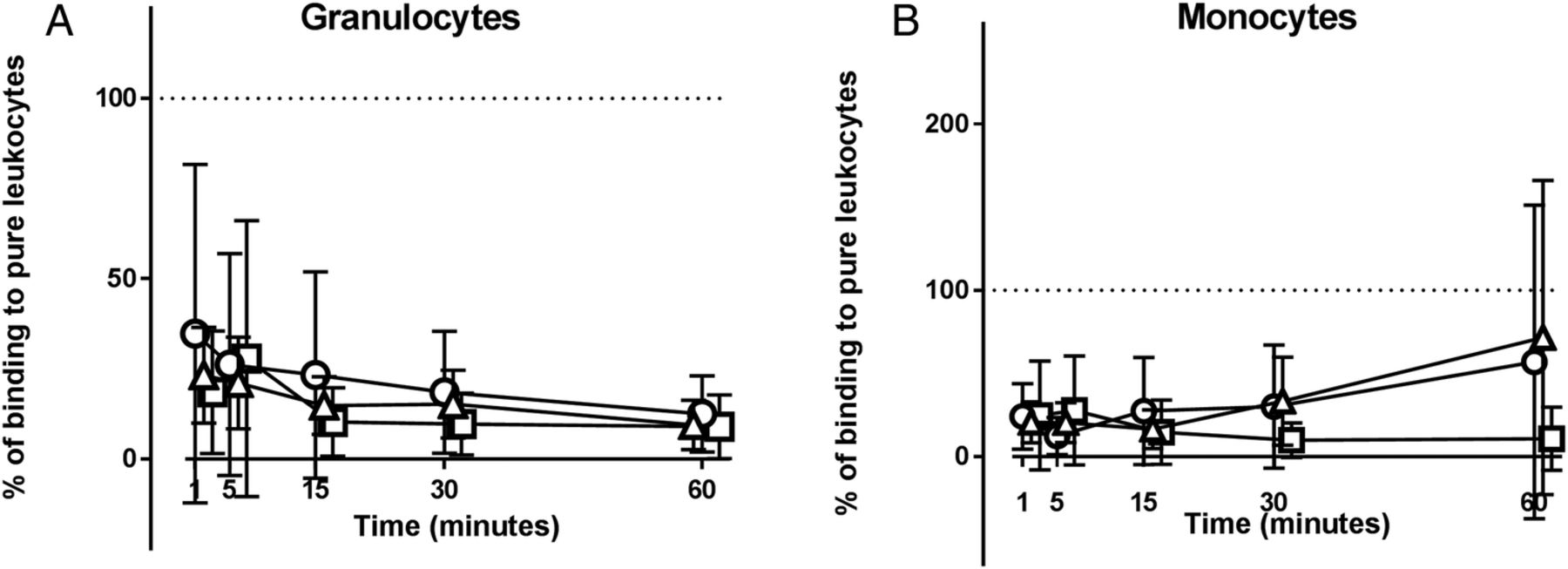
The presence of erythrocytes strongly inhibited binding of MPs to granulocytes and monocytes (figure 6). The inhibition of MP binding to granulocytes was statistically significant in patients with SLE and HCs at all time points, as was the binding to monocytes during the first 30 min of incubation. No differences were observed between HCs, patients with disease-active or patients with disease-inactive SLE in this respect (figure 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Erythrocyte-mediated inhibition of microparticle (MP) binding to phagocytes. Preparations of washed whole blood cells and purified leucocytes, from equivalent amounts of whole blood, collected from healthy controls (circles), patients with disease-active (triangles) and patients with disease-inactive (squares) SLE were incubated with carboxyfluorescein diacetate succinimidyl ester-labelled MPs for 60 min in the presence of 33% (v/v) autologous serum. The binding of MPs to (A) granulocytes and (B) monocytes contained in the preparations of washed whole blood cells is shown as percentage of the corresponding binding in the preparations of purified leucocytes (=100%, indicated by dotted line). Symbols and error bars represent mean and 95% CI. One experiment involving a healthy donor has been excluded due to lack of binding to all cell types.
Discussion
The main aim of this study was to investigate C3 fragments on MPs from patients with SLE and the ability of MPs to bind to circulating phagocytes and erythrocytes.
Few other studies have investigated the binding of complement to MPs. Here, we show that patients with SLE had a higher proportion of MPs with surface-bound C3 fragments than did HCs. In line with this, Mobarrez et al25 recently found fourfold to tenfold increased levels of platelet and endothelial cell-derived MPs expressing C4d in patients with SLE compared with HCs. Further, Østergaard et al11 demonstrated increased amounts of several complement proteins, including C3, on MPs from patients with SLE. We also found that MPs from patients with SLE carried diminished amounts of the opsonising C3b and iC3b fragments. The total amount of C3 in SLE serum was decreased, presumably as a result of increased complement C3 activation and consumption.6 Taken together, these findings indicate that the reduced amounts of complement C3 available in patients with SLE are distributed on a relatively larger proportion of MPs, presumably due to more antibodies on the surface of SLE MPs, ultimately resulting in diminished opsonisation of these MPs. This might be one explanation for the diminished phagocytosis of autoantigenic MPs described in SLE.7
Contributing to our finding of diminished complement C3b and iC3b fragments on MPs from patients with SLE may be the presence of C3b-blocking antibodies, as demonstrated by Kenyon et al.26 Reduction of opsonising signals on MPs from patients with SLE is further supported by the finding that patients with SLE have fewer annexin V-binding MPs, and thereby fewer MPs expressing phosphatidylserine on the surface, than do HCs.17 ,25 The diminished opsonisation of MPs in patients with SLE is likely to compromise the appropriate clearance of MPs by phagocytosis and via circulating erythrocytes, which would explain an increased tissue deposition of MPs, similar to that seen for ICs.27 ,28
The binding of MPs to granulocytes from patients with SLE was slightly increased. This might be explained by enhanced opsoninisation, especially with immunoglobulins, of MPs from patients with SLE, compared with MPs from HCs.5 Granulocytes from patients with SLE show decreased phagocytosis of dead cell material,8 but according to our findings, this is not related to diminished ability to take up MPs.
Here, we show that MPs bind equally well to erythrocytes from patients with SLE and HCs, and that this binding restricts the binding of MPs to circulating phagocytes, in the same manner as previously shown for ICs.29 Binding of MPs to erythrocytes has previously been demonstrated by Gasser and Schifferli,16 who found that neutrophil-derived MPs bound to erythrocytes in the presence of complement and that addition of anti-CR1 (3D9) completely abrogated this binding. However, the ability of erythrocytes to act as a buffer restricting MP binding to phagocytes is novel. We speculate that this buffering effect might also include restriction of MP presentation to antigen-presenting cells, thereby representing a mechanism to protect the immune system against autoantigens present on MPs.
Regarding MP binding to phagocytes, other research groups have shown that MPs generated in vitro from T cells and endothelial cells bound to monocytes.30–32 Our study adds to this by reporting for the first time on the binding of in vivo generated MPs to circulating phagocytes, and the potential differences in this binding between patients with SLE and HCs.
Platelet-derived MPs were the most abundant MP type in both HCs and patients with SLE, and this is consistent with earlier studies,17 ,25 though the percentage is not as high as what others have found (see online supplementary table). A lower percentage of platelet-derived MPs might be explained by the rapid centrifugation after sampling (30 min maximum), which diminishes MP release from platelets during storage. We found no differences between groups regarding levels of total MPs, or subfractions of annexin V-binding MPs; 7AAD-binding MPs; or platelet-derived, endothelial cell-derived or leucocyte-derived MPs. Other research groups have found varying differences in MP numbers and phenotype distribution—regarding cell of origin and membrane integrity—between patients with SLE and HCs, but in general, these studies demonstrate considerable variation in their results.17 ,19 ,25 ,33–37 It is possible that our study contributes to such variation by using fresh samples, as opposed to many other studies using frozen samples. Also varying use of filtering might play a role. Our finding of similar concentrations of total MPs in SLE and HCs may reflect that the supposedly compromised clearance of MPs in SLE38 is balanced by an increased tissue deposition.
This study contributes to the growing knowledge about mechanisms for clearing of MPs in SLE. Our findings indicate that the previously demonstrated diminished phagocytosis of MPs in patients with SLE39 is not due to diminished binding of MPs to phagocytes. However, our finding of reduced amounts of C3b and iC3b on the surface of MPs may lead to less efficient opsonisation and phagocytosis of MPs. Our demonstration of MP binding to erythrocytes supports the theory that MPs are also cleared by erythrocyte-mediated transportation to the liver, spleen or bone marrow, in parallel to the mechanism shown for ICs.6 ,16 ,40 ,41 Moreover, our finding that erythrocytes restrict the binding of MPs to granulocytes and monocytes indicates that erythrocytes buffer opsonised MPs, preventing their inappropriate activation of circulating phagocytes, as also shown for opsonised ICs.42 However, unlike previous findings for ICs,29 we did not find that erythrocytes from our SLE patients were less able to bind to MPs.
A limitation of this study is that we did not measure the amount of CR1 on the erythrocytes as our finding of the erythrocytes showing equal buffering capacity could be caused by equal numbers of CR1 on erythrocytes in the two groups. Another limitation is the lack of measuring the effect of adding anti-CR1 during the incubation—this would have increased our knowledge about mechanisms of MP binding to cells.
Future studies should include analysis of the mechanisms of MP binding to cells, especially concerning the role of the receptors involved. Studies are also warranted investigating the functional aspects of the binding of MPs to cells shown here, including differences between patients with SLE and HCs.
Acknowledgments
The authors thank Christoffer Tandrup Nielsen for sharing his experience in purifying microparticles with us. The authors also thank the patients for their participation, and the Department of Biostatistics at Copenhagen University for statistical guidance.
References
Footnotes
Contributors LKW designed the study, acquired the data, analysed and interpreted the data, and drafted the article. CHN designed the study, analysed and interpreted the data, and revised the manuscript critically for important intellectual content. SJ designed the study, interpreted data, and revised the manuscript critically for important intellectual content. All authors approved the final version of the article before submission.
Funding We thank The Danish Rheumatism Association, Gerda og Hans Hansens fond, Kong Christian den Tiendes fond, Overlæge Johan Boserup og Lise Boserups legat and Rigshospitalet for financial support.
Competing interests None declared.
Ethics approval The Danish Scientific Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.