Article Text

Abstract
SLE is characterised by an activation of the interferon (IFN) system, which leads to an increased expression of IFN-regulated genes. The reasons behind the IFN signature in SLE are (1) the existence of endogenous IFN inducers, (2) activation of several IFN-producing cell types, (3) production of many different IFNs, (4) a genetic setup promoting IFN production and (5) deficient negative feedback mechanisms. The consequences for the immune system is a continuous stimulation to an immune response, and for the patient a number of different organ manifestations leading to typical symptoms for SLE. In the current review, we will present the existing knowledge of the IFN system and pathway activation in SLE. We will also discuss how this information can contribute to our understanding of both the aetiopathogenesis and some organ manifestations of the disease. We will put forward some issues that are unresolved and should be clarified in order to make a proper stratification of patients with SLE, which seems important when selecting a therapy aiming to downregulate the IFN system.
- systemic lupus erythematosus
- interferon
- plasmacytoid dendritic cell
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE has for many years been a challenge to clinicians and a mystery for the basic scientist because of the complex clinical picture and the bewildering array of different aberrations in the immune system. Gradually, a number of observations in several research groups have unravelled important mechanisms behind the many clinical and laboratory findings, which now are translated into new therapies. However, several clinical trials have failed and there are a number of reasons for this. One cause is the fact that we still do not know how the genetic setup, environmental factors and stochastic events contribute to the initiation of the disease and the continuous autoimmune process. Obviously, several key elements of SLE need to be understood in more detail in order to completely unlock the secret behind the disease. Among key findings in SLE is a prominent expression of interferon (IFN)-regulated genes, an IFN signature, in blood and tissues.1–4 This observation was reported by several groups already in 2003 and initiated an intense activity among researchers trying to explain the finding. Simultaneously, colleagues started to investigate if the IFN signature could be linked to clinical phenotype, disease activity, comorbidities, treatment effects and prognosis. Even though much knowledge regarding the IFN system in SLE has been accumulated during the last 16 years, much is still unclear or unknown. For instance, what is the cause or trigger of the IFN signature? To what extent contribute type II and type III IFN, besides type I IFN, to the IFN signature? Which cells produce the IFN, and are different cells responsible for the IFN production during different phases of the disease? Shall we block the IFN system in SLE, and if so, which is the most suitable target? We want to bring forward some aspects that are important, not at least for the understanding of how to stratify patients when deciding on line of therapy.
Interferons
IFNs constitute a fundamental part of the defence against viral infections and were originally defined by their ability to ‘interfere’ with viral replication.5 During viral infections, large amounts of IFNs are produced, activating the antiviral machinery in IFN-exposed cells, resulting in inhibition of viral replication. Viruses try to evade this innate defence mechanism and one noticeable example was the ‘Spanish’ influenza that could block expression of multiple IFN-stimulated genes, which probably contributed to the high mortality in the 1918 pandemic.6
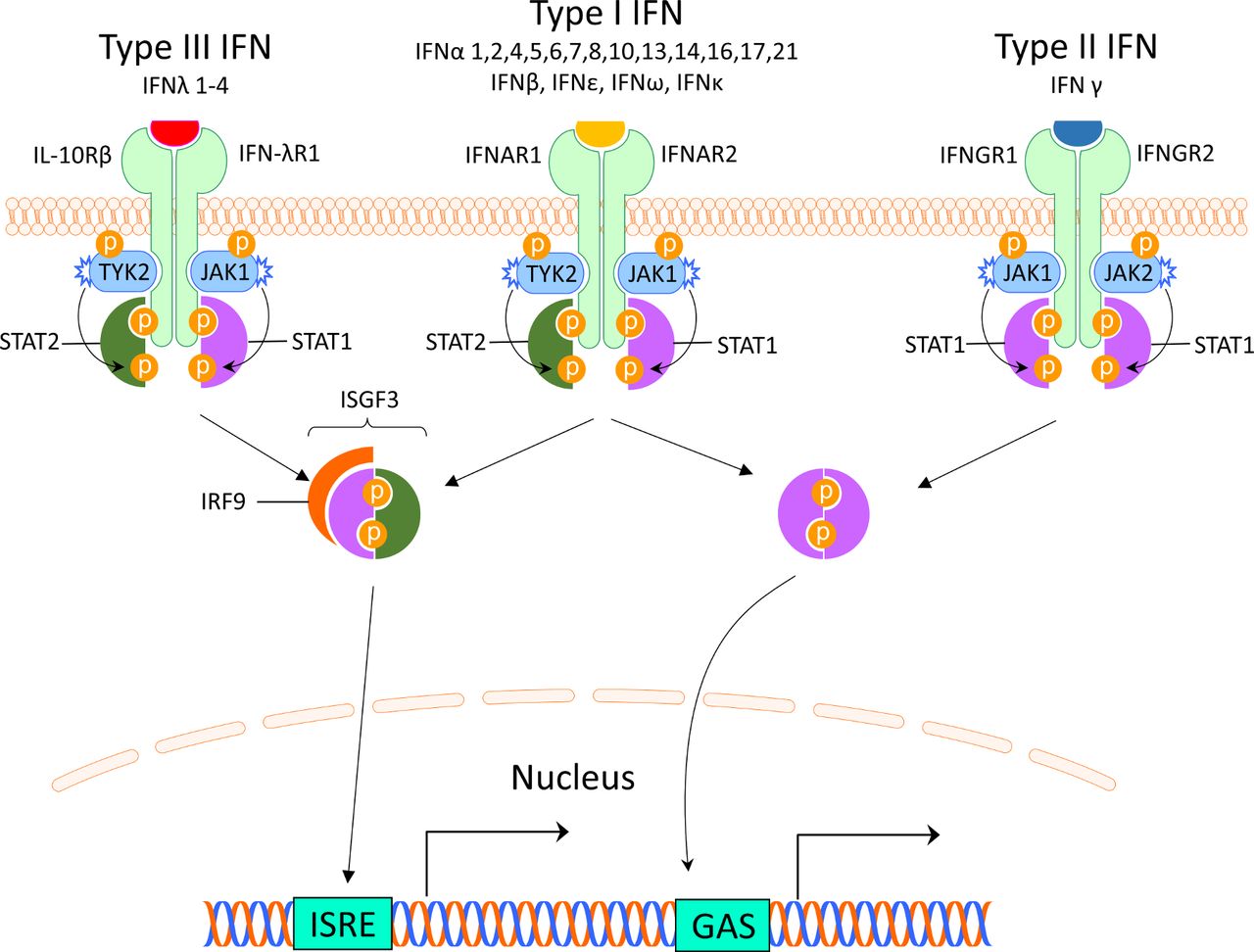
There are three different types of IFNs (I–III), and the type I IFNs are the largest family. It can be divided into five classes (IFN-α, β, ε, κ and ω), of which IFN-α can be further divided into 12 subtypes.7 Most cells can produce small amounts of type I IFN, but the principal type I IFN-producing cell is the plasmacytoid dendritic cell (pDC), originally called the natural IFN-producing cell.8 9 The type I IFNs are typically induced by viruses, bacteria or microbial nucleic acids when sensed by pattern recognition receptors (PRRs) localised in the cytosol or in the endosome. These PRRS include Toll-like receptors (TLRs), retinoic acid inducible gene 1 (RIG-I)–like receptors (RLRs) and nucleotide oligomerisation domain–like receptors (NLRs).10 Type I IFNs all bind to the same ubiquitously expressed type I IFN receptor (IFNAR) that consists of two polypeptide chains of IFNAR1 and IFNAR2. The subsequent signalling pathway involves activation of Janus kinase (JAK) 1 and tyrosine kinase (TYK) 2 and formation of the interferon-stimulated gene factor 3-complex (IGSF3), including signal transducer and activator of transcription (STAT) 1, STAT2 and interferon regulatory factor (IRF)9. IGSF3 binds to interferon stimulated response elements in promoters of IFN-regulated genes11 (figure 1).

Interferon receptors and signalling. The interferons are classified into three types, which bind to distinct receptors. This induces activation of overlapping pathways resulting in expression of different genes. GAS, interferon-gamma activated sequence; IFN, interferon; IFNAR, interferon alpha receptor; IFNGR, interferon gamma receptor; IFN-λR1, interferon lambda receptor 1; IL-10Rβ, interleukin-10 receptor β; IRF, interferon regulatory factor; ISGF3, interferon-stimulated gene factor 3; ISRE, interferon-stimulated response elements; JAK, Janus kinase; STAT, signal transducer and activator of transcription; TYK2, tyrosine kinase 2.
The type II IFN consists of only one member and many different cells produce IFN-γ including natural killer (NK) cells and T cells.12 IFN-γ binds to the IFN-γ receptor (IFNGR) which is expressed on most cells. Via activation of JAK1 and JAK2, ligation of the IFNGR results in phosphorylation of STAT1 homodimers and binding to IFN-γ-activated sites (GASs) and subsequent gene expression11 (figure 1). This signalling pathway can also be used by IFNAR and there is therefore a large overlap between type I and II induced genes.13
Type III IFNs comprise four newly identified lambda IFNs: IFNλ1/IL29, IFNλ2/IL28A, IFNλ3/IL28B and IFNλ4 (IFNL4).14 IFN-λs are most abundant at barrier surfaces including the respiratory and gastrointestinal tracts, and is produced by epithelial and epithelial-origin cells including hepatocytes and some immune cells (macrophages and DCs). The type III IFNs signal through a receptor complex (IFNLR1/IL10Rβ) that is primarily expressed on epithelial cells (gastrointestinal, respiratory and urogenital), hepatocytes and a few immune cells including neutrophils and DCs14 (figure 1).
Interferon in SLE
Increased levels of IFN in serum of patients with SLE was already described 40 years ago15 and were later identified as type I IFNs.16 Observational studies found that patients treated for malignancies with IFN-α could develop a lupus-like disease with autoantibodies to nuclear antigens, suggesting that type I IFN can break the tolerance and induce an autoimmune disease.17 18 When genome-wide expression analysis became available, several groups showed that 50%–75% of adult patients and up to 90% of children with SLE display an increased expression of type I IFN-regulated genes (an IFN signature).1–4 Younger patients have a more prominent IFN activity compared with older patients,19 and SLE disease activity correlates with IFN-α levels and the strength of the IFN signature.2 20–22 Analysis of longitudinal gene expression signatures suggest IFN-α to generate a relatively stable pattern of IFN-I transcripts over time, while other gene clusters may reflect induction of IFN-β or IFN-γ and present a more variable pattern.23 Thus, the IFN signature observed in patients with SLE probably corresponds to more IFN types than just IFN-α, although the type I IFN seems most important.
It is important to notice that a very large number of genes are regulated by IFNs and the specific genes expressed depend on the cell type, expressed receptors, type of stimuli and timing of sampling. Recent studies have suggested that perhaps 10% of our genes are regulated by IFN, but type III IFN induces a limited number of genes and no unique transcripts have been defined.24 Studies of the IFN signature in SLE have included different compositions of cells, often whole blood or peripheral blood leucocytes, and the number of genes examined have varied from only a few to large panels. There is also a significant overlap between the genes induced by type I, II and III IFN, which is why it has been difficult to differentiate among the IFNs contributing to the signature. The results have been inconsistent and sometimes challenging to interpret, as no consensus on how to measure the score exists today. However, all three IFNs seem to contribute to the signature.23 25 26 Perhaps not surprising, when comparing the IFN signature in patients with a viral infection and SLE, patients with SLE display a more complex pattern of gene expression.27 Studies of IFN expression in tissues have added further complexity to this field, as previous largely neglected IFNs may be important in specific organs, such as IFN-κ in cutaneous lupus.28
Triggers of interferon production in SLE
During the last years, a number of possible mechanisms explaining the persistent type I IFN production in SLE have been described, and in figure 2 several conceivable inducers of the IFN production are shown. An important mechanism of IFN-α induction is mediated via interferogenic ICs, which consist of autoantibodies and nucleic acid binding proteins.29–31 The ICs are endocytosed via FcγRIIa on pDCs, transported to the endosome where the nucleic acid part of the IC binds TLR7 or TLR9 with subsequent activation of transcription factors and IFN-α production32 (figure 3). This route of IFN induction has been demonstrated in vitro, combining purified SLE IgG and apoptotic or necrotic cell material as well as small nuclear ribonucleoproteins (snRNPs), which is relevant given the increased apoptosis and reduced clearance of apoptotic debris observed in patients with SLE.33 34

Inducers and regulators of IFN-α production by plasmacytoid dendritic cell. APC, antigen-presenting cell; GM-CSF, granulocyte-macrophage colony-stimulating factor; IC, immune complex; IFN, interferon; IL-3, interleukin 3; LFA1, lymphocyte function–associated antigen 1; MIP-1β, macrophage inflammatory protein-1β; NET, neutrophil extracellular traps; PECAM-1, platelet and endothelial cell adhesion molecule 1; ROS, reactive oxygen species.

Schematic picture of the type I interferon production and different nucleic acid sensors. cGAMP, cyclic GMP-AMP; cGAS, cyclic GMP-AMP synthase; DAI, DNA-dependent activator of IFN-regulatory factors; DDX41, DEAD-box helicase 41; DNA-PKcs, DNA-dependent protein kinase; ER, endoplasmatic reticulum; FCGRIIA, Fc-gamma receptor II A; IC, immune complex; IFI16, gamma-interferon-inducible protein 16; IRAK, interleukin-1 receptor–associated kinase; IRF, interferon regulatory factor; MAVS, mitochondrial antiviral-signalling protein; MDA5, melanoma differentiation–associated protein 5; MyD88, myeloid differentiation primary response 88; NET, neutrophil extracellular traps; RIG-I, retinoic acid–inducible gene I; STING, stimulator of interferon genes; TLR, Toll-like receptor; TRAF6, TNF receptor–associated factors; TREX1, three prime repair exonuclease 1.
NET formation is a cell death pathway where neutrophils extrude nuclear material such as histones, decondensed chromatin and cytoplasmatic proteins in a web-like structure. Patients with SLE have increased NET formation and an impaired capacity to degrade NETs due to decreased function of extracellular DNAse I.35 36 This increases exposure of nucleic acids and proteins to autoreactive B cells and autoantibodies, and it has been shown that NETs activate pDCs to produce high levels of IFN-α in a TLR9-dependent manner36 37 (figure 2). Recently, it was shown that SLE neutrophils extrude high levels of oxidised mitochondrial DNA that can induce IFN-α production via the cGAS stimulator of interferon genes (STING) pathway.38 Furthermore, NET-derived cationic antimicrobial peptide LL37–DNA complexes expand self-reactive memory B cells to produce anti-LL37 Abs in an Ag-dependent manner.39 Highlighting the importance of exposure of autoantigens to the immune system for IC formation and IFN induction, other described self-derived IFN inducers include high mobility group box chromosomal protein and heat shock protein 90.40–42
Transposable elements (TA) are DNA sequences that can change position within a genome and constitutes more than half of the human DNA.43 It represents a potential significant source of stimulatory self-nucleic acid. Long interspersed nuclear element-1 (L1) is a class I TA and 80–100 L1s are thought to remain active in any given individual.43 Hypomethylation of L1 has been observed in SLE and is associated with increased L1 expression.44 45 In kidney biopsies of patients with SLE nephritis, increased expression of L1 has been observed and correlates with expression of type I IFN.45 L1 triggers IFN production by pDCs via TLR7 but also by monocytes via cytoplasmic RIG-I.45
Infections can trigger the onset of SLE or a disease flare, and although many viruses have been implicated in the aetiology of SLE, no specific virus or bacteria has been identified to cause the disease. Gut microbiota can activate STING and induce type I IFN,46 and recently it was shown that the Gram-positive bacteria Enterococcus gallinatum translocate across the gut barrier and induce Th17 and Tfh cells as well as innate immune pathways including the pDC/IFN axis.47 Treatment of the lupus-prone mice (NZW×BXSB)F1 with antibiotics inhibited autoantibody production and ameliorated disease. Given the impaired gut barrier function observed in SLE, these data highlight the possibility that the gut microbiome could be of aetiopathogenetic importance in at least a subset of patients with SLE.
In conclusion, there exist a large number of possible inducers of IFN production in SLE and probably different inducers are most important in different patients. Greater understanding of the relevant trigger(s) and pathways mediating the IFN production in individual patients would be of great help in order to develop precise treatments that target the specific IFN inducers causing a persistent IFN production.
Interferon-producing cells in SLE
The number of pDCs is reduced in the circulation of patients with SLE, but can be detected in inflamed tissues, such as skin48 49 and kidneys, where they seem to be activated.50 Several lines of evidence suggest that pDCs to a great extent are responsible for the ongoing IFN production in SLE. Thus, in murine models of lupus, depletion of pDC ameliorates the disease51 and genetically impaired pDC function improves the disease.52 A recent study also showed that targeting pDC in patients with SLE decreases the expression of IFN response genes in blood, reduces cutaneous immune cell infiltrates and ameliorates skin lesions.53 Despite these observations, the role of other IFN-producing cells in SLE needs to be clarified because a number of studies suggest that several other cell types are involved in IFN production. Among these are the keratinocytes, which can produce both IFN-κ28 and IFN-λ26 and monocytes, which have been implicated in the generation of the IFN signature.54 Monocytes are mainly responsible for IFN production in the pristaine-induced murine lupus model, which is characterised by a prominent IFN signature, and human monocyte-derived macrophages transfected with a small non-coding Y RNA or stimulated with immune complexes express IFN-α and IFN-β mRNA.55 However, the precise role of IFN-producing monocytes in human SLE is at the moment unresolved and needs to be further explored. Neutrophils have the capacity to produce type I IFN56 and bone marrow–derived neutrophils in patients with SLE produce IFN-α.57 Produced IFN seems to promote alterations in B-cell development with a reduction in the fraction of pro/pre-B cells, suggesting an inhibition in early B-cell development and an expansion of B cells at the transitional stage. This could well be an early event in the breakage of tolerance and development of autoimmunity with autoantibody production. Besides type I IFN-producing cells, activated NK cells in SLE have an increased production of IFN-γ.58 Furthermore, detectable IFN-λ transcripts have been noted in peripheral blood leucocytes from patients with SLE,59 but the exact source of IFN-λ in patients with SLE is at the moment unclear.
An important observation is that several cell types, once activated, can stimulate pDC to an increased IFN production. Thus, NK cells, B cells and T cells all can enhance IFN production when pDCs are exposed to nucleic acid–containing immune complexes60–62 (figure 2). The in vivo relevance of these findings remains to be established, but suggests that in SLE there is an extensive cross-talk between different immune cells and pDCs, which promotes the ongoing IFN production and sustained autoimmune process.
In summary, several cell types can contribute to the IFN signature seen in patients with SLE, and although pDC most probably is the main source of the IFN, it seems conceivable that in a subset of patients, other cell types are important IFN producers that need to be targeted in order to completely control the activated IFN system.
Genetic factors influencing the IFN system in SLE
Today, more than 100 genetic risk loci have been associated to SLE63 and more than half of the identified SLE susceptibility genes encode proteins with functions directly or indirectly linked to type I IFN production or responses.64 65 These include genes involved in Toll-like receptor activation and their downstream signalling molecules. For most of the risk gene variants, the mechanism by which the risk gene contributes to disease susceptibility, or severity, is unknown, but recent studies have shed some light on this issue. One of the strongest SLE risk loci outside the HLA region is signal transducer and activator of transcription (STAT)4, which has been known as a SLE risk locus for more than 10 years.66 The intronic SLE-associated STAT4 SNPs are linked to a disease phenotype with an earlier onset and an increased risk for stroke and nephritis with severe renal insufficiency.67–69 We recently showed that activated T cells from STAT4 risk allele carriers with SLE have increased levels of STAT4 protein, resulting in more phosphorylated STAT4 in response to IL-12 and IFN-α, and an augmented IL-12-induced IFN-γ production.70 In contrast, activated T cells from healthy individuals carrying the STAT4 risk allele displayed a decreased response. This finding may be of importance as to why the majority of risk allele carriers do not develop disease and suggests that the STAT4 risk allele needs to interact with other host or environmental factors to be pathogenic. As pre-incubation of healthy donor cells with IFN-α selectively enhanced the IL-12 response in STAT4 risk allele carriers, it is possible that STAT4 risk allele carriers are at risk to develop SLE during prolonged type I IFN production.71 These results also link a SLE susceptibility locus to both the type I and type II IFN systems, and one could speculate that these findings at least partly can explain the good therapeutic response to the anti-IL-12/IL-23 mAb ustekinumab in a proportion of patients with SLE.72
The interferonopathies comprise a group of rare monogenic diseases with a constitutive overproduction of type I IFN caused by mutations in genes responsible for handling of nucleic acids.73 Thus, these patients have various disturbances in the intracellular nucleic acid metabolism or in cytosolic nucleic acid–sensing pathways, which cause an autoimmune or autoinflammatory disease. The patients have a prominent IFN signature, but show a remarkable phenotypic heterogeneity, which indicates that other genes and environmental factors modify the inflammatory response. Some of the patients have a clear SLE phenotype, and it is possible that genes responsible for the interferonopathies also contribute to the development of the disease in a subset of patients with SLE normally encountered at the rheumatology department. In fact, a recent study of whole-genome sequencing of patients with SLE shows that ultra-rare, coding heterozygous variant connected to the diverse spectrum of interferonopathies are over-represented among patients with SLE.74
Epigenetic changes are prominent in cells and tissues from patients with SLE and a recent, comprehensive review summarises DNA methylation studies in SLE.75 Patients with SLE have decreased methylation levels in a large number of CpG sites and the most strongly hypomethylated genes are IFN regulated.76 Furthermore, in a study of twins discordant for SLE, there was a higher degree of hypomethylation in IFN-regulated genes among twins that had experienced a flare within the past 2 years, which link the epigenetic IFN signature to a more active disease.77 Since some epigenetic changes are mitotically heritable and relatively stable, it is possible that some of the demethylated sites in IFN-regulated genes are hypomethylated already in utero, or received from one of the parents, although IFN exposure during disease flares seems most plausible.
Taken together, genetic studies demonstrate that the genetic risk for development of SLE is strongly connected to gene variants in the IFN signalling pathway and changes in IFN-regulated genes. The mechanisms by which these alterations are involved in the development of SLE are under intense studies, but results so far strengthen the assumption that the genetic setup directly contributes to an IFN-driven autoimmune process.
Connection between the IFN system and other immune cells
As mentioned above, a number of cells in the immune system can interact with pDC and enhance the IFN response. Perhaps even more important are the effects of produced IFN on most cells in both the innate and adaptive immune systems (reviewed in Eloranta et al8). Type I IFN acts as an immune adjuvant and one mechanism for the enhanced immune response by type I IFN is an increased expression of MHC I molecules,78 which facilities the cross-presentation of exogenous antigens as well as detection of virus-infected cells by cytotoxic T cells. IFN also promotes the expression of a number of other molecules important in the immune response, such as MHC II, CD40, CD80 and CD86, but also the expression of chemokines and their cognate receptors such as CXCL10 and CXCR3. DCs stimulated by IFN mature and achieve a molecular repertoire that makes them very potent as antigen-presenting cells and capable to induce differentiation of naïve CD4+ T cells, but also development of CD8+ memory T cells. Type I IFN increases the differentiation of Th17 cells and suppress Treg functions, which can all lead to an expansion of autoreactive T cells79
Type I IFNs impact B-cell function through a variety of mechanisms that lead to prolonged survival and activation, including differentiation and class-switch recombination causing enhanced antibody production (reviewed in Kiefer et al80). Type I IFNs increase the production of B-cell activating factor in monocytes and via this mechanism stimulate antibody production.81 NK cells are considered important in at least a subgroup of patients with SLE82 and type I IFNs increase the cytotoxicity and IFN-γ production in NK cells, linking both the type I and type II IFN system to each other.83
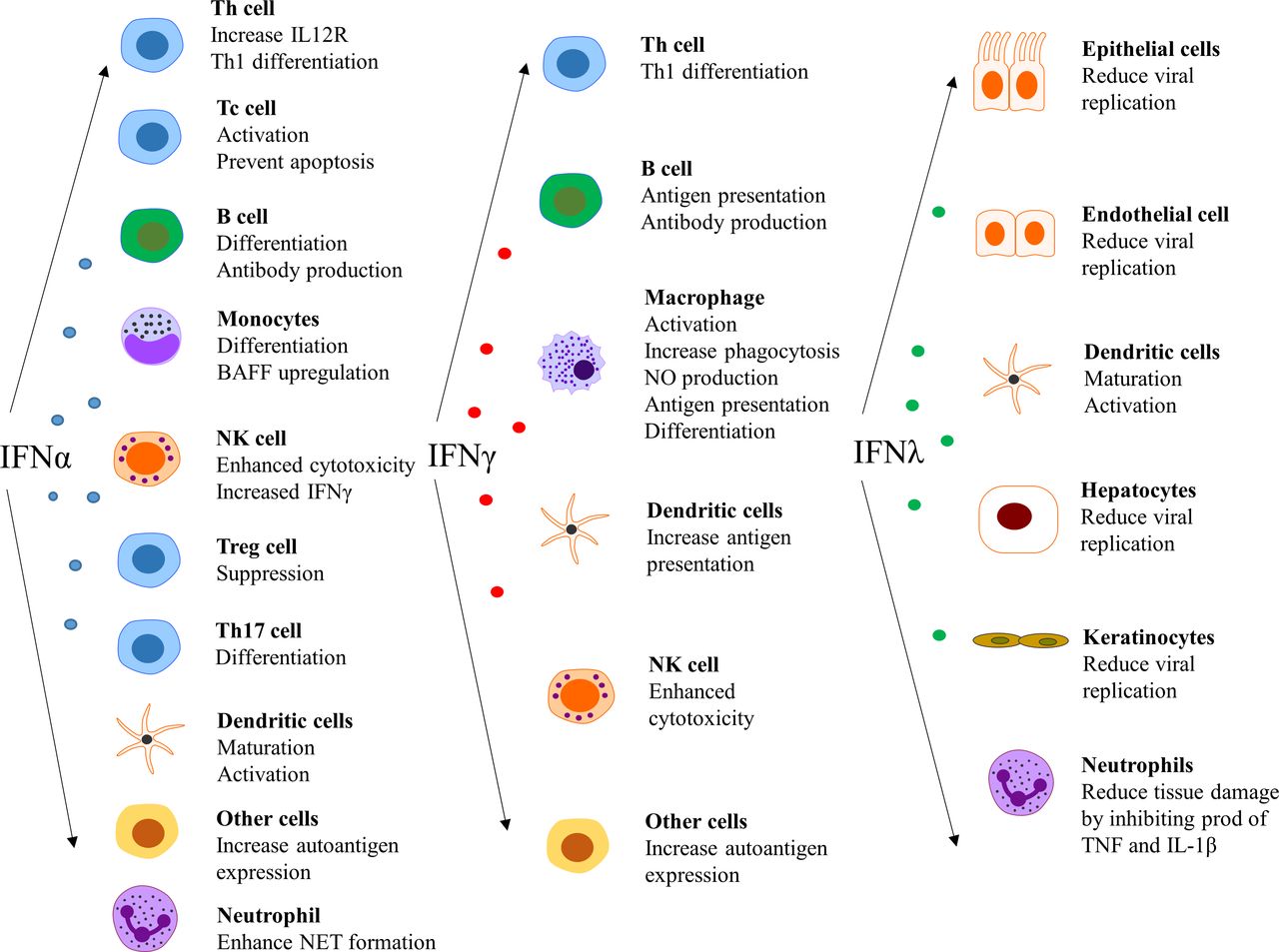
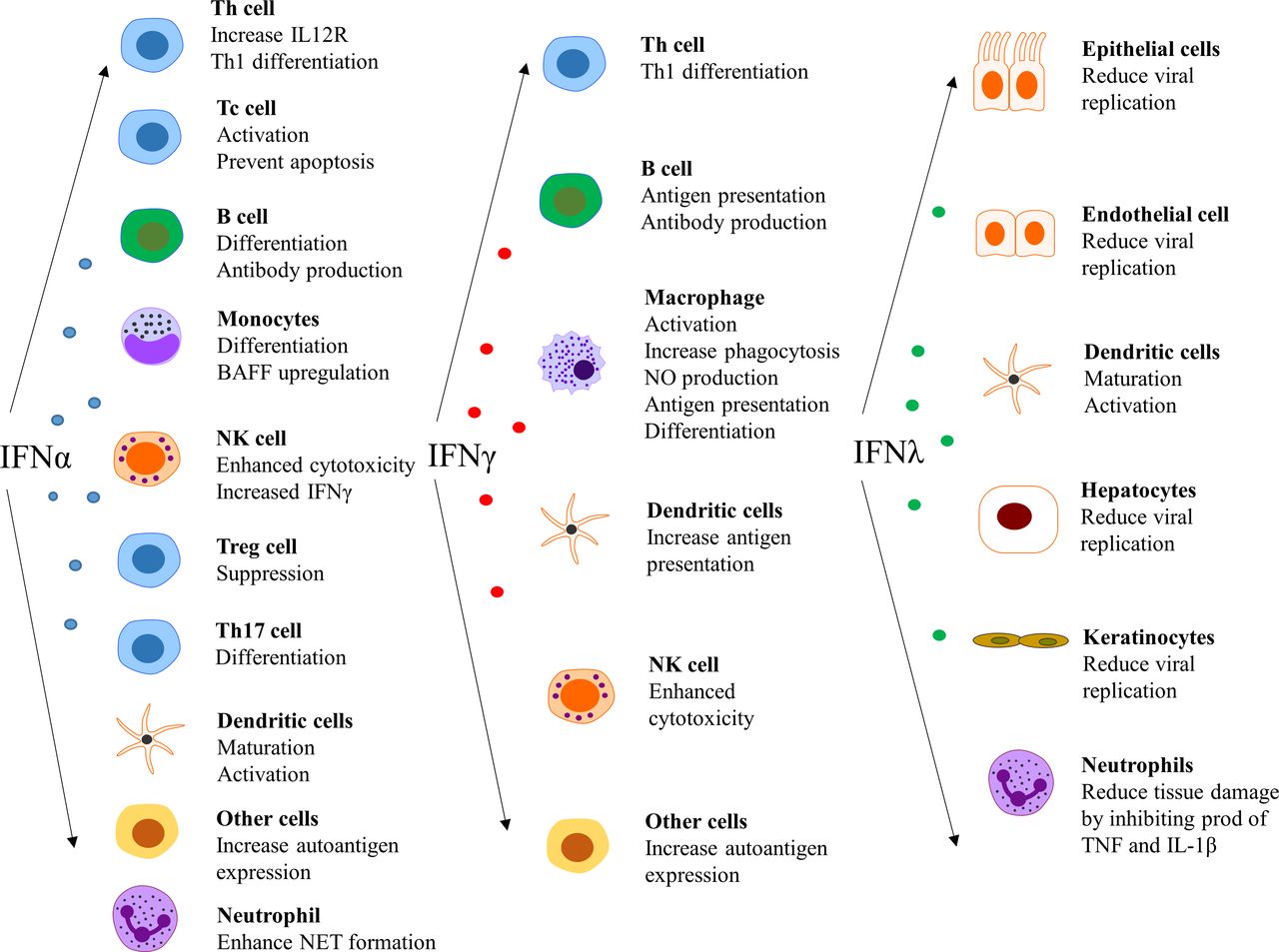
The type I IFNs also have effects outside the immune system, such as impairment of endothelium-dependent vasorelaxation and endothelial progenitor cell function, which slow down the repair process of damaged endothelium.84–86 Further, IFN-α inhibits eNOS expression and impairs insulin-mediated nitric oxide production in endothelial cells,87 enhances foam cell formation88 and alters platelet function.89 These observations can be linked to the unexpectedly high prevalence of atherosclerosis and cardiovascular disease in patients with SLE.90 In a more general perspective, it is clear that there are several detrimental effects on cells and tissues that are exposed to IFN for an extensive period of time.91Figure 4 summarises the effect of various IFNs on different cell types.14 25 92 93
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of interferons (IFNs) on different cell types. NET, neutrophil extracellular traps.
Disease process in SLE
The many findings concerning the IFN system in patients with SLE can be put together into an aetiopathogenic model of SLE, which has been reviewed elsewhere.94 For instance, an initial infection by a virus can induce type I IFN production and release of cellular material from dying cells. Several other triggers of IFN production also exist, as discussed above. The extracellular autoantigens from apoptotic and necrotic cells as well as NETs from granulocyte then trigger B cells to autoantibody production against RNA and DNA binding proteins in individuals prone to autoimmune reactions. ICs will be formed, which act as endogenous type I IFN inducers, causing a prolonged stimulation of type I IFN production by pDCs. The excessive release of endogenous DNA/RNA in combination with impaired clearance of apoptotic cell material will facilitate the production of IFN by a number of cell types. This will result in chronic activation of the IFN system, which will drive an autoimmune process leading to chronic inflammation and tissue damage in a vicious circle manner.
IFN system and disease manifestations
A number of signs and symptoms in patients with SLE are connected to the increased production of IFN. General symptoms of acute viral infections such as muscle and joint pain, headache, pleurisy, fatigue and fever are associated with type I IFN.20 IFN also has a suppressive effect on the bone marrow resulting in anaemia, neutropenia, lymphopenia and thrombocytopenia,95 and consequently a high IFN signature score has been associated with the haematological American College of Rheumatology criteria.2 Presence of anti-dsDNA, anti-SSA and anti-RNP is associated with high IFN-α activity96 and antibodies to RNA with a strong IFN signature,21 highlighting the link between IFN and B-cell maturation and autoantibody production.
Skin
Patients with hereditary interferonopathies often present with cutaneous manifestations including malar rash and alopecia.97 In SLE, the IFN signature correlates with cutaneous disease activity98 and IFN-regulated genes are expressed in epidermis and dermis of cutaneous lesions.28 99 Increased expression of IFN-λ126 and IFNκ28 has been observed in the skin of patients with cutaneous SLE and type I IFN has been suggested to inhibit ADAM17 in Langerhans cells thereby enhancing UVB-induced keratinocyte apoptosis.100 101 Recent phase I–II trials in SLE blocking IFN signalling via pDC,53 IFNAR102 or the JAK/STAT pathway103 have all improved skin manifestations and JAK inhibition has also improved skin manifestations in patients with interferonopathies.104 Thus, IFN signalling seems to be a key player in SLE skin pathology even though the exact interplay between different IFNs, keratinocytes and pDCs needs further exploration.
Arthritis
Synovial tissue from patients with lupus with inflammatory arthritis demonstrate increased expression of IFN-induced genes,105 and recent data suggest IFN signatures induced by IFN-β1 and IFN-α2 to be most important.106 The source of IFN in the synovium is not clear, but fibroblast-producing IFN-β, which are rich in this tissue, have been suggested.106 107 Results from the Anifrolumab trial, blocking IFNAR, show significant improvement of joint disease in patients with lupus with an initially strong IFN signature, indicating an important role for IFNs in lupus arthritis.102
Kidneys
Regarding SLE nephritis, studies have shown an association between a strong IFN signature and both a history of nephritis2 21 and active nephritis.22 Kidney biopsies of patients with SLE show increased expression of IFN-inducible genes108–110 and pDCs accumulate in glomeruli of patients with active disease.50 A stronger IFN signature is observed in glomeruli and renal tubule in patients with immunosuppressive naïve SLE with nephritis compared with patients with SLE nephritis having received immunosuppressive treatment.111 In kidney tissue, IFN-β can induce podocyte cell death and increase permeability whereas both IFN-α and IFN-β suppress renal progenitor cell differentiation into mature podocytes,112 resulting in podocyte loss, proteinuria and impaired glomerular repair. Taken together, these observations suggest that IFN is important in both the inflammatory process and development of damage in SLE nephritis.
Central nervous system
Increased levels of type I IFN have been demonstrated in the cerebrospinal fluid of patients with SLE with neuropsychiatric manifestations,113 including lupus psychosis114 and also in the central nervous system (CNS) post mortem.114 This is intriguing given the observed adverse neuropsychiatric effects following IFN-α treatment.115 Locally produced cerebrospinal fluid autoantibodies from patients with CNS lupus can form ICs and stimulate IFN-α production by pDCs116 and type I IFN stimulates microglia to become reactive and engulf neuronal and synaptic material in lupus-prone mice.117 Thus, it is possible that interferogenic ICs and type I IFN can be of importance for the neuropsychiatric (NP) manifestations often observed in SLE and NP-SLE may be one manifestation of the disease suitable for IFN inhibition.
Targeting the IFN system
After the discovery of the IFN signature, a number of different strategies have been developed in order to downregulate the IFN system in patients with SLE. So far, the therapeutic effect has been modest and difficult to reproduce in larger phase III studies.118 There are several reasons for this, but there are a number of factors that need to be taken into consideration before selecting therapeutic target in a patient with SLE. Several have been discussed above and some are summarised in table 1.
Factors to consider before selecting the therapeutic target in a patient with SLE
Recent clinical trials have stratified patients by clinical manifestations, including nephritis or skin and joint manifestations. Unfortunately, several trials have failed, which is why in the selection of patients, the molecular pathways activated in a single patient must also be taken into consideration. In this context, it is important to note that the type I IFN system may be most critical early in the disease process2 18 119 120 and at initiation of flares.20 Later in the disease course, other IFN subtypes may have a more prominent role, at least in some patients.72 Therefore, defining the IFN and pathway activation of importance for individual patients will be necessary. This analysis also includes the many pathways related to the IFN system. Attempts have been made to refine the IFN signature using factor analysis and by linking ISG expression to IFN subtype.23 121 Others have applied single-cell RNA sequencing of lupus kidney biopsies, identifying a high IFN response signature in tubular cells as a negative prognostic marker of lupus nephritis.122 Further studies of how specific cells and cell types contribute to the disease process and organ manifestations may reveal how to personalise the treatment for the patients and also on the cellular level.
Genetic profiling will also help to determine the underlying mechanism of disease in single patients. Individuals with rare monogenic SLE, including patients with rare variants of genes linked to interferonopathies,74 or genetic complement deficiency may benefit from individualised treatment. As discussed above, patients with risk gene variants linked to IFN-γ signalling including STAT4 and IL12 might benefit from inhibition of this pathway. In the future, it will perhaps be important to consider the cumulative genetic risk, as defined by a genetic risk score, when selecting therapy as this may predict disease outcome.123 In the years to come, we expect that genetic studies (genotype–phenotype) will give us more information about what pathways to target in the individual patient.
Conclusion
The IFN system is our most fundamental defence system against infections, but in patients with SLE, there is an ongoing production of IFN that sustains an autoimmune process. The complexity of the IFN system, together with the many clinical features of SLE, has made it difficult to target the proper molecules in single patients. However, during the last years, we have seen a dramatic increase in the understanding of the IFN system and its role in SLE. Although this information has added more elements to consider in our clinical decision process, we are now closer than ever to unlock the mystery of how to target the IFN pathway in SLE.
Acknowledgments
We would like to acknowledge the critical review of the manuscript by Niklas Hagberg and Maija-Leena Eloranta.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
Footnotes
Contributors LR and DL wrote and approved the manuscript.
Funding The study was supported by the Swedish Rheumatism Association, King Gustaf V’s 80-years Foundation, the Swedish Research Council and the Swedish Society of Medicine (the Ingegerd Johansson donation).
Competing interests LR has received a research grant from AstraZeneca and received honoraria for scientific advice from Biogen.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Data availability statement No additional data are available.