Article Text

Abstract
Background/objective Treatment with immune checkpoint inhibitors (ICIs) in oncology patients is increasing. Although ICIs trigger rheumatic immune-related adverse events, development of SLE features has been rare. Whether long-term treatment with ICIs would promote SLE features remains unknown. To begin to address this, we generated SLE-prone NZM 2328 mice with lifelong reduction in CTLA-4 expression.
Methods Since CTLA-4-deficient (Ctla4− /−) NZM mice developed a lethal lymphoproliferative disorder by 3–6 weeks of age, development of SLE in these mice could not be studied. Ctla4 haploinsufficient NZM.Ctla4+ / − mice were assessed in parallel with littermate female NZM.Ctla4+ / + mice. Evaluations included CTLA-4 expression and lymphocyte profiles, assessed by fluorescence-activated cell sorting; serological profiles, assessed by ELISA; renal immunopathology, assessed by histology and immunofluorescence; and clinical courses, assessed by mortality.
Results CTLA-4 expression was lower in NZM.Ctla4+ / − mice than in NZM.Ctla4+ / + mice. Spleen mononuclear cells, B cells, plasma cells, CD4+ cells, recently activated CD4+ cells and CD4+ T regulatory (Treg) cells were increased in NZM.Ctla4+ / − mice (p≤0.042). The serological profile, degree of renal immunopathology and mortality in NZM.Ctla4+ / − mice remained unaffected.
Conclusion Lifelong reduction in CTLA-4 expression in NZM mice neither accelerated nor aggravated SLE. Expansion in Treg cells may have played a protective role. Our observations raise the hope that long-term treatment of patients with SLE with an anti-CTLA-4 agent, should the need arise, would not adversely affect SLE disease activity.
- CTLA-4
- lupus
- animal model
- checkpoint protein
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune checkpoint proteins, including CTLA-4, are important contributors to immunological tolerance. In most resting T cells, CTLA-4 expression is limited,1 whereas CTLA-4 is expressed constitutively by T regulatory (Treg) cells.2 3 Mice genetically deficient in CTLA-4 (Ctla4− / − mice) developed lethal lymphoproliferation by 3–6 weeks of age,4 5 and treatment of human CTLA4 knock-in mice with an anti-CTLA-4 mAb promoted development of circulating anti-dsDNA antibodies.6 Partial blockade of CTLA-4 promoted development of juvenile-onset diabetes in mice that bore a type-1 diabetes-permissive MHC locus (H2g7),7 and administration of anti-CTLA-4 mAb accelerated and exacerbated onset and severity of experimental autoimmune encephalomyelitis and autoimmune diabetes.8 9
Nevertheless, whereas rheumatic immune-related adverse events (IRAEs) have been well documented in human oncology patients treated with immune checkpoint inhibitors (ICIs), including the anti-CTLA-4 mAb ipilimumab (reviewed in Calabrese et al 10), only a single case of de novo SLE has been reported.11 Only 1 of the 16 patients with an already-diagnosed systemic rheumatic disease treated with ICIs at the Mayo Clinic exhibited a flare of the underlying rheumatic disease, and neither of the two patients with SLE treated with ICIs flared.12 Importantly, none of the 61 ICI-treated patients who developed an IRAE developed SLE features.13 An important caveat, however, is that treatment durations with ICIs have been short, so longer term consequences of ICI treatment on SLE remain to be established.
To investigate whether a long-term reduction in CTLA-4 expression could affect SLE features, we introduced Ctla4 haploinsufficiency into SLE-prone NZM 2328 (NZM) mice and assessed the consequences on the development and course of SLE. Although the lymphocyte profile of NZM.Ctla4+ / − mice differed from that of littermate NZM.Ctla4+ / + wild type (WT) mice, the lifelong reduction in CTLA-4 expression neither accelerated nor aggravated SLE disease. This raises the hope that long-term treatment of patients with SLE with ipilimumab (or other anti-CTLA-4 agents), should the need arise, would not adversely affect SLE disease activity.
Materials and methods
General
All reported studies were approved by the USC IACUC.
Mice
All mice were housed in a single specific pathogen-free room. NZM.Ctla4+ / − mice were generated by introgressing the Ctla4+ / − genotype from B6.Ctla4+ / − mice14 into NZM wild-type (WT) mice.15 The N7 backcross generation was fully congenic.
As with non-autoimmune-prone Ctla4− / − mice,4 NZM.Ctla4− / − mice develop a lethal lymphoproliferative syndrome by 3-6 weeks of age (unpublished observations), so development of SLE could be assessed only in NZM WT and NZM.Ctla4+ / − mice. Accordingly, NZM.Ctla4+ / − mice were mated with NZM WT mice, giving rise to pups, 50% being Ctla4+ / + and 50% being Clta4+ / − . Since development of SLE in NZM mice is female predominant (as in humans), only female mice were studied.
Cell surface staining
Spleen mononuclear cells were stained with combinations of fluorochrome-conjugated mAb specific for CD3, CD4, CD8, CD19, CD25, CD44, CD62L, CD69, surface immunoglobulin (Ig)D or surface IgM (BioLegend or BD Bioscience) to identify total B (CD19+) cells, follicular (FO) B (CD3−CD19+IgMlo/−IgDhi) cells, marginal zone (MZ) B (CD3−CD19+IgMhiIgDloCD1dhi) cells, plasma (CD3−CD138+CD19lo) cells, total CD4+ cells, total CD8+ cells, activated memory CD4+ cells and recently activated CD4+ (CD4+CD69+) cells. Data were analysed with FlowJo V.7.6.1 software (Treestar).
Intracellular staining for CTLA-4
Spleen mononuclear cells were surface stained for CD4 and CD25, fixed and permeabilised and stained with Alexa Fluor 488-conjugated anti-Foxp3 mAb and phycoerythrin (PE)-conjugated anti-CTLA-4 mAb. Control samples were identically treated, substituting PE-conjugated Armenian hamster IgG isotype control mAb for the anti-CTLA-4 mAb (BioLegend). Cells were gated on the CD4+CD25+Foxp3+ (Treg cell) population, and data were analysed for CTLA-4 expression.
Serum total IgG and IgG anti-dsDNA levels
Serum levels of total IgG and IgG anti-dsDNA were determined by ELISA.15 Anti-dsDNA antibody OD values were normalised to the mean OD of serum from 5-month-old MRL-lpr mice, the latter arbitrarily assigned a value of 100 U/mL.
Kidney histology
Sections of formalin-fixed kidneys were stained with H&E and assessed by light microscopy.15
Kidney immunofluorescence
Sections of snap-frozen kidneys were stained for IgG or C3 deposition using fluorescein isothiocyanate-conjugated goat F(ab′)2 fragment anti-mouse IgG or C3 antibodies (MP Biomedicals).15
Assessment of clinical disease
Since the USC IACUC requires euthanisation of moribund mice or mice with >20% wt loss, mice were often euthanised before they could develop fixed severe proteinuria (≥3+ by dipstick). Accordingly, the clinical endpoint was the age of natural death or the age at which the mouse was compassionately euthanised.
Statistical analysis
All analyses were performed using SigmaStat software (SPSS). Parametric testing between two groups was performed by the unpaired t-test. When the data were not normally distributed or the equal variance test was not satisfied, non-parametric testing was performed by the Mann-Whitney rank sum test between two groups. Survival data were analysed by the log-rank test.
Results
Expression of CTLA-4 in NZM.Ctla4 + / − and NZM WT (Ctla4 + / + ) mice
NZM.Ctla4− / − mice reproducibly developed a lethal lymphoproliferative syndrome by 3–6 weeks of age (unpublished observations) indistinguishable from that developed by CTLA-4 deficient C57BL/6 (B6) or BALB/c mice.4 5 That is, mice genetically deficient in CTLA-4 bearing a SLE-prone genetic background developed the same syndrome as did mice genetically deficient in CTLA-4 bearing a non-autoimmune-prone genetic background. Accordingly, only NZM.Ctla4+ / − and NZM WT mice survived to adulthood.
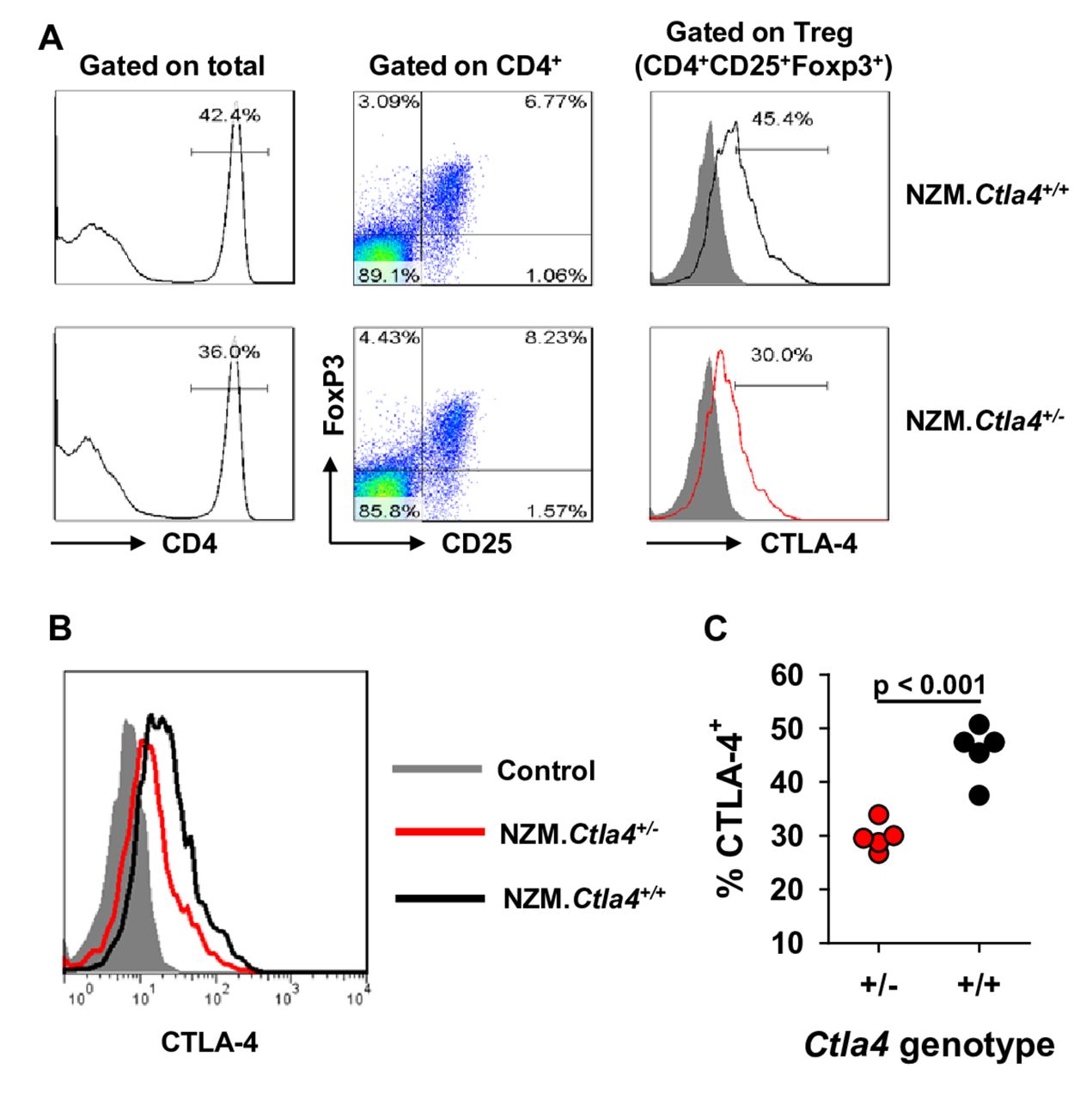
To demonstrate that the CTLA-4 phenotype corresponds to the Ctla4 genotype, expression of CTLA-4 in Treg cells from 2-month-old NZM.Ctla4+/− and littermate NZM WT mice was assessed. CTLA-4 expression was uniformly lower in Treg cells from the former than from the latter (figure 1A,B), thereby resulting in fewer CTLA-4+ Treg cells harboured by NZM.Ctla4+ / − mice than by NZM WT mice (figure 1C). Confirmatory experiments with littermate 3-week-old B6 WT, B6.Ctla4+ / − and B6.Ctla4− / − mice bearing a Foxp3-gfp knock-in documented the absence of CTLA-4 in B6.Ctla4− / − mice, with B6.Ctla4+ / − +/− expressing CTLA-4 at an intermediate level relative to that expressed by B6 WT mice (online supplementary figure 1).
Supplemental material

CTLA-4 expression in Treg cells from NZM.Ctla4 + / − and NZM WT mice. (A) Spleen cells from littermate 2-month-old NZM.Ctla4+ / − and NZM.Ctla4+ / + female mice were stained for surface CD4, surface CD25, intracellular Foxp3 and intracellular CTLA-4. The cells were gated on the CD4+ population (left) and then on the CD25+Foxp3+ population (middle). CTLA-4 expression by the T regulatory (Treg) (CD4+CD25+Foxp3+) cell population is shown on the right. CTLA-4 staining intensity is plotted on a logarithmic scale. Results from representative NZM.Ctla4+ / + and NZM.Ctla4+ / − mice are shown. (B) CTLA-4 tracings from another representative NZM.Ctla4+ / − mouse (red) and another representative NZM.Ctla4+ / + mouse (black) are superimposed to highlight the difference in staining intensities. (C) Composite results from five female NZM.Ctla4+ / − mice (red circles) and five female NZM.Ctla4+ / + mice (black circles) are plotted. Each circle represents an individual mouse.
Lymphocyte profiles
By 2–3 months of age, differences in lymphocyte profiles between NZM.Ctla4+ / − and NZM WT mice were appreciated. Spleen mononuclear cells, B cells, FO B cells, plasma cells, CD4+ cells, activated memory CD4+ cells, recently activated CD4+ cells and Treg cells were all greater in Ctla4+ / − mice than in Ctla4+ / + mice (figure 2A). No differences were observed in MZ B cells (median 2.16×106 vs 1.98×106, p=0.403) or CD8+ cells (mean 15.3×106 vs 12.2×106, p=0.243). In addition, no difference in CD4 mean fluorescence intensity (MFI) of CD4+ cells was observed (normalised MFI 100.0%±4.4% for NZM WT mice vs 99.5%±3.6% for NZM.Ctla4+ / − mice; p=0.677).

Lymphocyte and serological profiles of NZM WT and NZM.Ctla4 +/− mice. (A) Spleen mononuclear cells, B (CD19+) cells, follicular B (CD3−CD19+IgMlo/−IgDhi) cells, plasma (CD3−CD19loCD138+) cells, CD4+ cells, CD4+ activated memory (CD4+CD44+CD62L−) cells, CD4+ recently activated (CD4+CD69+) cells and T regulatory (CD4+Foxp3+CD25+) cells from NZM.Ctla4+ / − (+/−; N=11) and NZM WT (+/+; N=9) mice aged 2–3 months are plotted. Results are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 10th and 90th percentiles. Each circle represents an individual mouse. (B) Sera from NZM WT (N=7–18 mice per age; circles) or NZM.Ctla4+ / − mice (N=7–22 mice per age; diamonds) at the indicated ages were assayed for total IgG (left) and IgG anti-dsDNA antibodies (right). Results are shown as medians, and the error bars indicate 25th and 75th percentiles. *p<0.05; **p<0.01. Ig, immunoglobulin; WT, wild type.
Serological profiles
The difference in lymphocyte profiles between NZM.Ctla4+ / − and NZM WT mice notwithstanding, the serological profiles of these mice were remarkably similar. Serum total IgG levels were highly variable in both cohorts, and the only time point at which a significant difference in median serum IgG levels was appreciated was at 5 months of age (figure 2B, left).
Serum IgG anti-dsDNA antibody levels also showed considerable variability. Substantial serum IgG anti-dsDNA antibody levels were detectable in both NZM WT and NZM.Ctla4+ / − mice by 2 months of age and reached their peaks at 5 months of age (figure 2B, right). IgG anti-dsDNA antibody levels tended to decline in older mice, although this decline likely was apparent rather than real, since earlier mortality among mice with high serum IgG anti-dsDNA antibody levels precluded availability of their sera at older ages. As with serum total IgG levels, serum IgG anti-dsDNA antibody levels were similar in both cohorts, with a significant difference in median values observed only at 7–8 months of age.
Clinical disease and renal immunopathology
In line with their similar serological profiles, mortality in the two cohorts was similar (p=0.581; figure 3A). At 5–7 months of age (the age at which the mice began to become clinically sick), similar degrees of glomerular deposition of IgG and C3 were evident in both sets of mice, accompanied by histological evidence of glomerulosclerosis, fibrotic crescents, interstitial inflammation and fibrosis and tubular atrophy and plugging (figure 3B).

{kind=link}
{kind=link}
{kind=link}
Clinical disease and renal immunopathology in NZM wild-type (WT) mice and NZM.Ctla4 + / − mice. (A) NZM WT (NZM.Ctla4+ / + ) mice (N=29) and NZM.Ctla4+ / − mice (N=43) were monitored for survival. (B) Kidney sections from NZM.Ctla4+ / + and NZM.Ctla4+ / − +/− aged 5–7 months were stained for immunoglobulin (Ig)G immunofluorescence or C3 immunofluorescence or with H&E for histological evaluation. Representative sections are illustrated. Original magnifications: 100× for immunofluorescence; 200× for H&E.
Discussion
Genetically imposed lifelong reduction of CTLA-4 expression did not alter serologic profile, degree of renal immunopathology or mortality in NZM mice. The expansion in B cells, plasma cells, activated memory CD4+ cells and recently activated CD4+ cells in NZM.Ctla4+ /− mice may have been counterbalanced by the concurrent expansion in Treg cells. To complement the genetic approach taken in the present manuscript, future studies will be undertaken in NZM WT mice treated long-term with anti-CTLA-4 antibodies. Such pharmacologically based studies would serve as a vital confirmation of the present genetic-based studies.
Studies in which the Ctla4 gene had been conditionally ablated in adult mice support the notion of homeostatic upregulation of Treg cells.16 These mice remained clinically healthy without evidence of lymphoproliferation or autoimmunity and their activated memory CD4+ cells and Treg cells were each expanded. Of note, IL-10 production by both Treg cells and T conventional cells was increased in these mice as was expression of the inhibitory molecules, Lag3 and programmed cell death protein-1 (PD-1). It may be that subtotal loss of CTLA-4 (as would occur following pharmacological neutralisation of CTLA-4) in adulthood, by maintaining a balance between pathogenic effector cells and protective regulatory cells, would neither promote autoimmunity de novo nor aggravate an underlying autoimmune state. We are currently generating NZM.Ctla4+ / − mice with a Foxp3-gfp knock-in so that unperturbed Treg cells can be isolated and directly analysed for their in vitro functions as well as their in vivo role in the non-acceleration of disease.
To date, seven ICIs have been approved by the FDA: the anti-CTLA-4 mAb, ipilimumab; the anti-PD-1 mAbs, nivolumab and pembrolizumab and the anti-PD-L1 mAbs, atezolizumab, avelumab, durvalumab and cemiplimab. Although de novo SLE nephritis in an ipilimumab-treated patient has been reported,11 no de novo cases of SLE have yet been reported in patients treated with the other ICIs. Moreover, neither of the two patients with SLE treated at the Mayo Clinic with ICIs experienced a flare in their disease.12 Although there is no reason to believe that patients with SLE patients would be less susceptible to treatment-related IRAEs than patients without SLE, one may be cautiously optimistic that long-term pharmacologic blockade of CTLA-4 will not aggravate SLE disease per se. ICI treatment could be offered to a patient with SLE with an ICI-responsive tumour without first trying other (less effective) treatment modalities. Given that ICI-based therapy now frequently combines the targeting of CTLA-4 with the targeting of PD-1/PD-1L, studies that address the effect of long-term reduction of PD-1 and/or PD-1L (with and without concurrent reduction of CTLA-4) on SLE disease are warranted.
References
Footnotes
Contributors WS designed the study and wrote the first draft. NY performed most of the experiments. SC performed some of the experiments. All authors analysed the data, contributed intellectually to the final draft and approved the final version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.