Article Text

Abstract
Background SLE is associated with increased risk of diffuse large B-cell lymphoma (DLBCL). DLBCL is routinely classified by cell of origin (COO), with germinal centre B-cell (GCB) being more common and indicating better prognosis in the general population. We studied COO subtyping in patients with SLE diagnosed with DLBCL and their survival.
Patients and methods We evaluated 20 cases of SLE with DLBCL. Immunohistochemistry analysis was performed (BCL2, MYC, BCL6, CD10, CD20, FOXP1, GCET1, MUM1) in tissue microarrays. We examined associations between molecular and clinical features, including overall survival.
Results Of the 20 DLBCL SLE cases, 12/20 cases (60%) were classified as non-GCB using Hans or Choi algorithms. MYC and BCL2 protein expression was positive in 6/20 (30%) and 8/20 (40%) SLE cases, respectively, with 2/20 (10%) co-expressing both markers. Seven (7/20) had only extranodal involvement at DLBCL diagnosis. As expected, non-GCB cases had worse survival. Cases presenting exclusively with extranodal disease were associated with shorter SLE duration and better survival despite higher BCL2 protein expression.
Conclusions We present novel data characterising DLBCL in SLE. Sixty per cent of the DLBCL in patients with SLE were non-GCB. The nodal and extranodal distribution in SLE was similar to what is known in the general population, but extranodal disease occurred more often with short SLE duration and was associated with longer overall survival. More research on cancer in SLE is the key to further understanding the complex interplay between cancer and the immune system.
- systemic lupus erythematosus
- diffuse large b-cell lymphoma
- epidemiology
- immunohistochemistry
- cell-of-origin
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- systemic lupus erythematosus
- diffuse large b-cell lymphoma
- epidemiology
- immunohistochemistry
- cell-of-origin
Introduction
It is well recognised that patients with SLE are at increased risk of developing haematological malignancies.1 2 In particular, non-Hodgkin’s lymphoma (NHL) is clearly increased in patients with SLE compared with the general population.3 As in the general population, the majority of NHL in patients with SLE are diffuse large B-cell lymphomas (DLBCLs).2
DLBCL has been stratified by gene expression profiling into two major groups associated with their cell of origin (COO).4 The germinal centre B-cell-like (GCB) subtype is more common and associated with better patient outcomes; it is related to BCL2 gene rearrangements and PI3K/AKT/mTOR pathway activation along with EZH2 gain-of-function mutations.5 6 The non-GCB type is mostly composed of lymphoma cells with an activated B-cell profile. Non-GCB DLBCL have a worse prognosis and are associated with nuclear factor-kappa B (NF-κB) and Janus kinase and signal transducer and activator of transcription proteins (JAK–STAT) pathway activation. Such pathways are typically activated by mutations of cluster of differentiation 79A/B (CD79A/B), caspase recruitment domain family, member 11 (CARD11), tumour necrosis factor alpha-induced protein 3 (TNFAIP3 or A20) and myeloid differentiation primary response gene 88 (MYD88).7–12
Subtyping of DLBCL is now required by the WHO guidelines and part of routine care in most institutions, as it plays an important role in prognosticating the outcome of patients and guiding management.13 14 It is unknown to date if the worse survival observed in DLBCL in patient SLE compared with the general population is proportionally driven by both main subtypes or one in particular. The use of immunohistochemistry (IHC)–based algorithms (most commonly the Hans algorithm) is an acceptable alternative to gene expression profiling in the determination of COO in DLBCL.6 15
SLE is a unique model to study the development and clinical behaviour of lymphoproliferative malignancies such as DLBCL. Our objective was to describe a series of DLBCL in patients with SLE, and study associations between clinical and histopathological features.
Subjects and methods
We studied patients with SLE3 16 with DLBCL occurring any time after SLE diagnosis and if formalin-fixed, paraffin-embedded tissue blocks were available. All cases fulfilled the American College of Rheumatology criteria and the diagnosis was also clinically confirmed by tertiary centre rheumatologists. The specimens were submitted to Vancouver, BC and reviewed by two pathologists (PF and BT-C). Twelve cases were interpreted from tissue microarray slides, built using two 0.6 mm cores per case from areas of tumour as determined by routine microscopy on H&E-stained sections. The other eight cases were reviewed on 4 µm whole-tissue slides. Clinical information including both date of SLE and DLBCL diagnosis were available for each case of DLBCL.
IHC markers for pathology review by PF and BT-C included CD10 (clone 56C6; Ventana Medical Systems, Tucson, Arizona, USA), BCL6 (clone LN22; Dako, Glostrup, Denmark) and MUM1 (MUM1p; Abcam, Cambridge, UK), GCET1 (clone RAM431; Abcam), FOXP1 (clone JC12; Thermo Fisher Scientific, Waltham, Massachusetts, USA), BCL2 (clone 124; Dako) and MYC (clone Y69; Ventana Medical Systems). In order to subclassify the DLBCL cases by COO, IHC was performed using the same methods and cut-off values as described by Hans et al17 (CD10 ≥30%, BCL6 ≥30%, MUM1 ≥30%) and Choi et al (GCET ≥80%, MUM1 ≥80%, CD10 ≥30%, BCL6 ≥30%, FOXP1 ≥80%), the two most commonly used COO determination IHC algorithms.6 MYC and BCL2 positivity were defined as staining in over 40% and 50% of tumour cells, respectively, and co-expression of both proteins was termed dual expressers.18
Subsequent analysis was performed using results from the Hans algorithm to facilitate comparison with other studies. With overall survival (ie, time until death) as the outcome variable, Kaplan-Meier survival curves were used to visualise differences in survival with respect to COO and nodal status. Significance for Kaplan-Meier analysis was determined using the log-rank test. We assessed if key demographic or clinical features were significantly associated with either GCB versus non-GCB or nodal versus extranodal disease. These variables included (1) age at DLBCL diagnosis (years), (2) age at SLE diagnosis (years), (3) SLE duration (years), (4) sex (female=reference), (5) presence of extranodal involvement (any nodal involvement=reference), and expression (percentage of positive cells) of (6) BCL2, (7) BCL6 and (8) MYC.
Univariable Cox proportional hazards models were performed for (1) sex, (2) year of SLE diagnosis (to control for treatment modality), (3) nodal status, (4) COO and (5) age of cancer diagnosis. At the risk of overfitting the data, a final adjusted multivariable model using all variables was tested. All analyses were performed in R (V.3.3.3) using RStudio (V.0.99.902) with add-on data.table (V.1.10.4) and survival (V.2.41) packages.
Results
We analysed 20 cases of DLBCL for which tumour tissue was available; 12 cases were from Sweden, seven from Canada and one from the USA. Of these, 18 (90%) were female, the median age at the time of lymphoma diagnosis was 58 (range 33–82, average 56) years and the median SLE duration at DLBCL diagnosis was 11 (range 1–29, average 12) years. Seven of 20 (35%) DLBCLs had only extranodal involvement at initial diagnosis, 10 (50%) had only nodal involvement and 3 (15%) had both nodal and extranodal disease. Table 1 summarises the demographic and clinical characteristics and provides IQRs where appropriate.
Descriptive features of the SLE–DLBCL (n=20) cohort
The IHC analysis showed CD10 positivity in 6/20 (30%), BCL6 6/20 (30%), MUM1 7/20 (35%), GCET1 0/20 (0%), FOXP1 7/20 (35%) and CD20 20/20 (100%). MYC IHC was positive in six (30%) and eight (40%) were positive for BCL2, two of which were dual expressors (MYC and BCL2). Twelve cases (60%, 95% CI 39% to 78%) were classified as non-GCB whereas 8/20 (40%, 95% CI 22% to 61%) were classified as GCB. Except for two divergent cases, the two algorithms showed identical results. Of the eight GCB cases, three had extranodal involvement (one with both nodal and extranodal disease) compared with seven (two with both nodal and extranodal) in the 12 non-GCB cases.
The median survival for all 20 cases was 39 (mean 64) months. For the GCB cases, median survival was 67 (mean 103) months and for the non-GCB cases median survival was 17 (mean 38) months. Median survival was 86 (mean 106) months for patients with extranodal involvement only, 28.5 (mean 48.3) months in patients with nodal involvement only and 8 (mean 17.3) months in cases with both nodal and extranodal disease.
Stratified Kaplan-Meier survival curves suggested that both non-GCB subtype and nodal status were associated with worse overall survival (figure 1). In both univariate and multivariable Cox proportional hazards models, both non-GCB type and nodal status (any nodal involvement vs none) were associated with lower survival (table 2).

Kaplan-Meier analysis of overall survival based on (A) cell of origin and (B) nodal status. GCB, germinal centre B-cell.
Cox proportional HR for survival in SLE–DLBCL (n=20)
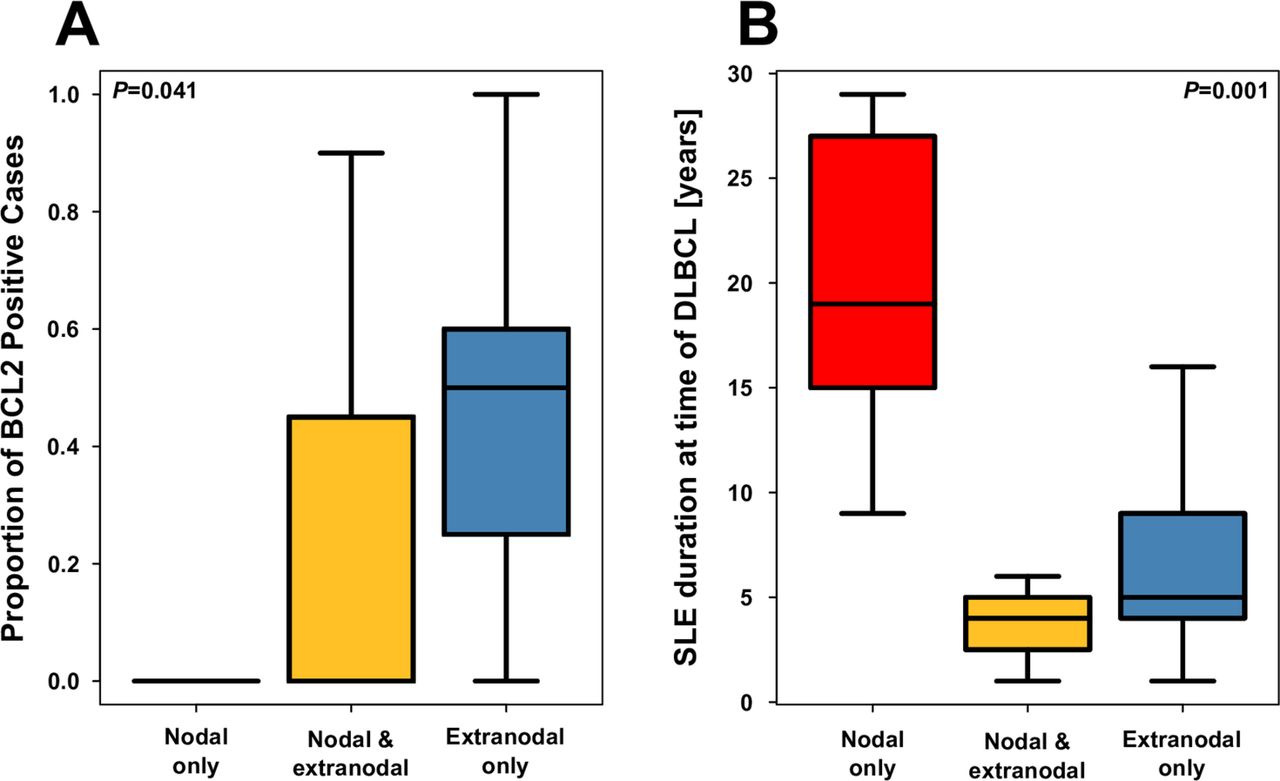
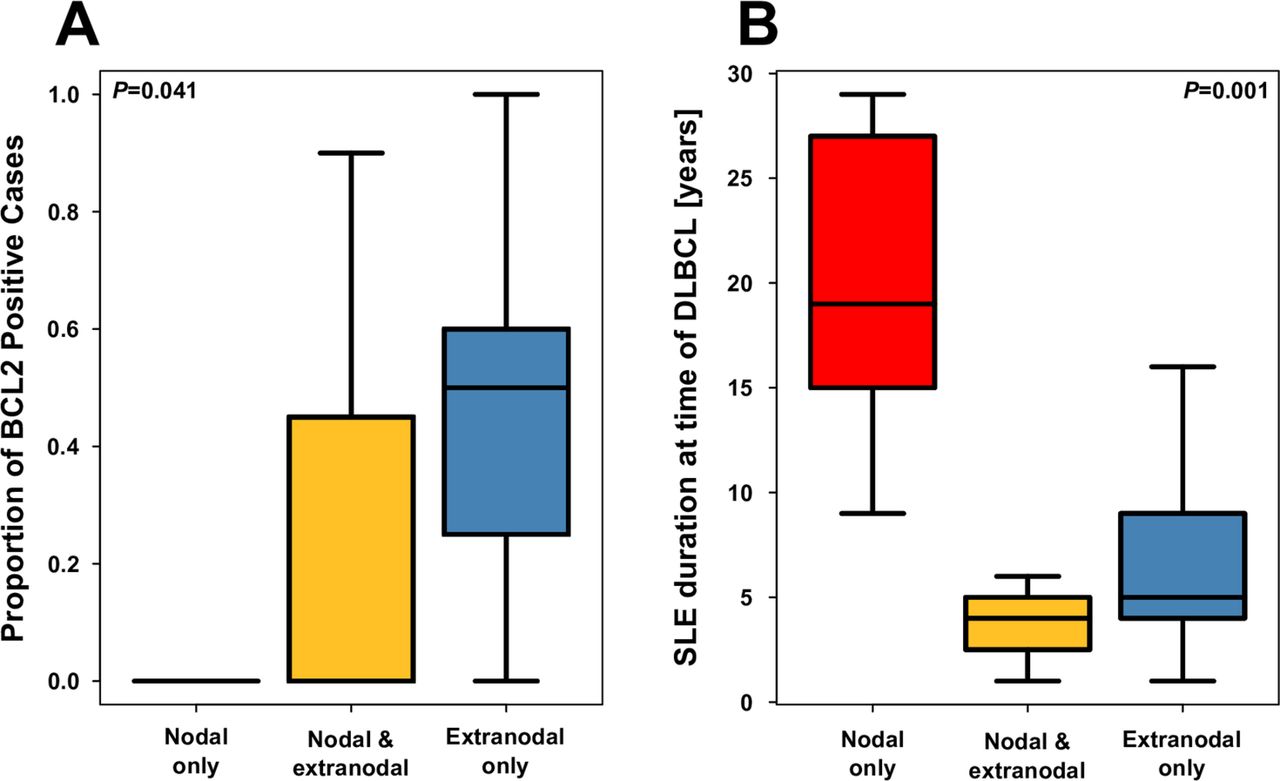
Stratification of cases by nodal status revealed that SLE duration at DLBCL diagnosis was much longer in cases presenting with nodal-only involvement (figure 2) and that BCL2 expression tended to be greater in patients presenting with extranodal involvement only. We were unable to detect any molecular or clinical features differing by COO subtype.
{kind=link}
{kind=link}
Box plot comparing cases of nodal and extranodal disease in terms of (A) proportion of BCL2-positive cells and (B) SLE duration at the time of diffuse large B-cell lymphoma (DLBCL).
Discussion
We and others have described a more than fourfold increase in lymphoma for patients with SLE compared with the age-matched and sex-matched general population.3 The reason for this is not fully understood, but both immunological abnormalities (which may be driven by genetic factors) and drug exposures (primarily cyclophosphamide) may play a role.19–21
Sixty per cent of the DLBCL in patients with SLE were non-GCB. In recent analyses of a population-based registry study of DLBCL (n=348) using the Hans algorithm, 41% were non-GCB and 59% were GCB.22 Our SLE sample, with 60% of cases being non-GCB, certainly tends towards the upper limit of what has been described in the general population. As in the general population, the non-GCB subtype in SLE cases was associated with shorter survival. The rate and pattern of incidence of dual expressers of MYC and BCL2 in our cohort is comparable with the general population, where dual expressors tend to be non-GCB types and are associated, in the general population, with poorer survival.23 24 One of these dual expresser SLE cases with disease localised to the lymph node had short survival (3 months) whereas the other had extranodal involvement only and a much longer survival (85 months).15 18 It must be acknowledged that the interpretation of our results is limited by the sample size. However, taken together, these results may suggest that the effect of SLE on overall DLBCL incidence and survival is unlikely to be specific for the biology of either subtype, as discussed in the following paragraph.
From a pathophysiological perspective, SLE itself potentially shares mechanisms with both GCB and non-GCB types. Non-GCB DLBCLs rely on the activation of the NF-κB and JAK–STAT pathways, both directly affected in SLE through derangements of A20, tumour necrosis factor superfamily (TNFSF4), TNF-α, CD79, CARD11 and interleukin-1 receptor-associated kinase 1 (IRAK1) activity as well as epigenetic modifications.21 25–29 Interestingly, in primary Sjögren’s syndrome, an autoimmune rheumatic disease at high risk for mucosa-associated lymphoid tissue (MALT) lymphoma, most MALT cases have either germline polymorphisms of TNFAIP3, related to the A20 protein important in NF-κB activation, or somatic alterations of the gene within the lymphoma tissue.30 Moreover, polymorphisms of TNFAIP3 are common to rheumatoid arthritis (yet another condition linked with lymphoma) and Hodgkin’s lymphoma.31 In previous genome-wide association analyses, our group was unable to confirm a strong relationship with the lupus-related TNFAIP3 single-nucleotide polymorphism (SNP) rs7749323 specifically for DLBCL, but this may be a sample size issue. In those analyses, the rs2205960 SNP, related to TNFSF4, was associated with an OR per risk allele of 1.07, 95% CI 1.00 to 1.16, p value 0.0549.32 The OR for the SLE interferon regulatory factor risk allele rs12537284 (chromosome 7q32, gene) was 1.08, 95% CI 0.99 to 1.18, p value 0.0765.
The STAT4 lupus risk SNP rs7582694 meanwhile was not clearly linked to DLBCL. Our interpretation is that TNFAIP3, TNFSF4 and possibly interferon pathways are of high interest as potential mediators of the risk of DLBC (particularly non-GCB type) in SLE. However, it will be interesting to see if emerging SLE treatments based on JAK kinase inhibition could ultimately modulate the risk of DLBCL in lupus populations.33
On the other hand, DLBCLs of the GCB type are often defined by PI3K/AKT/mTOR pathway which may be hyperactivated in SLE secondary to defective PTEN expression.5 6 34 Other evidence showed that cyclophosphamide, a medication used to treat lupus, is partly responsible for the increased incidence of lymphomas in patients with SLE, but it is unclear if it would favour development of a particular molecular subtype.35 Rapamycin and other mTOR inhibitors have the potential to block this central pathway in both autoimmune diseases like SLE and also various forms of cancer. The advent of this new approach in SLE represents another way that novel drug development could potentially modify some of the altered cancer risk in lupus.
Just over half of our DLBCL cases were diagnosed at a SLE duration beyond 10 years. Extranodal involvement was more common in patients with shorter SLE duration, and those cases with only extranodal involvement had the best survival. Those with both nodal and extranodal involvement had the worst survival, with nodal-only cases showing intermediate results. This is somewhat different than what is seen from population-based studies where extranodal involvement is typically a marker for worse survival.36 It is unclear why extranodal involvement of DLBCL (despite being also associated with BCL2 protein expression, itself a marker of poor prognosis22 37) would be associated with better survival, but the results may be driven by the association with shorter SLE duration (although our previous analyses suggested higher all-cause standardised mortality ratios in low-duration SLE). The results may alternatively reflect a type of detection bias (if, eg, extranodal DLBCL in SLE is picked up earlier than nodal-only disease). However, extranodal DLBCL presenting symptoms are often more subtle than nodal DLBCL, where systemic B-symptoms are common,38 often triggering investigation.
In general, chronic inflammation is associated with lymphoproliferative disorders and could establish an environment fertile to the development of DLBCL in both nodal and extranodal sites.39 We note that the ratio between nodal and extranodal involvement does not seem to be different between patients with SLE and the general population.
Summary
Sixty per cent of the DLBCL in patients with SLE were non-GCB. We believe that TNFAIP3, TNFSF4 and possibly interferon pathways are of high interest as potential mediators of the risk of DLBC (particularly non-GCB type) in SLE. The nodal and extranodal distribution was similar between patients with SLE and the general population, but extranodal disease occurred more often in patients with short SLE duration and was associated with longer overall survival. Although sample size limits the interpretation of our results, our findings suggest that the immunological alterations in patients with SLE influence the tumour biology of DLBCL. More research studying cancer in SLE will be key to understanding the complex interplay between cancer and the immune system, especially as emerging lupus treatments could have important effects on related pathways.
Supplemental material
Acknowledgments
We thank Dr Björn Löfström for his help supervising the case collection.
References
Footnotes
Presented at Preliminary data on this work were previously presented at the American College of Rheumatology 2018 Annual Meeting. Abstract number 710 (https://acrabstracts.org/abstract/an-analysis-of-cell-of-origin-in-diffuse-large-b-cell-lymphoma-in-systemic-lupus-erythematosus-including-molecular-and-clinical-factors-associated-with-survival/).
Contributors SB, AEC, BT-C and PF designed the research study. PF, RG, EB, NJ, CB, DLK, AEC, RR-G, JL and SB contributed essential clinical data and tumour tissue. BT-C and PF performed the pathology review and immunohistochemistry scoring. BT-C, PF, DT and SB analysed the data. All authors contributed to the interpretation of results and writing of the manuscript. All authors approved the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The study was approved by the Research Ethics Board of the McGill University Health Centre (GEN-06-031) and the participating institutions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available upon reasonable request.