Article Text

Abstract
Type I interferons (IFN) can have dual and opposing roles in immunity, with effects that are beneficial or detrimental to the individual depending on whether IFN pathway activation is transient or sustained. Determinants of IFN production and its functional consequences include the nature of the microbial or nucleic acid stimulus, the type of nucleic acid sensor involved in inducing IFN, the predominant subtype of type I IFN produced and the immune ecology of the tissue at the time of IFN expression. When dysregulated, the type I IFN system drives many autoimmune and non-autoimmune inflammatory diseases, including SLE and the tissue inflammation associated with chronic infection. The type I IFN system may also contribute to outcomes for patients affected by solid cancers or myocardial infarction. Significantly more research is needed to discern the mechanisms of induction and response to type I IFNs across these diseases, and patient endophenotyping may help determine whether the cytokine is acting as ‘friend’ or ‘foe’, within a particular patient, and at the time of treatment. This review summarises key concepts and discussions from the second International Summit on Interferons in Inflammatory Diseases, during which expert clinicians and scientists evaluated the evidence for the role of type I IFNs in autoimmune and other inflammatory diseases.
- systemic lupus erythematosus
- interferon
- autoimmune diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Type I interferons (IFN) are a major line of host defence against viruses and other microorganisms.1 However, it is now clear that type I IFNs can also drive context-specific responses to infection, which may be either beneficial or detrimental to the host.2–4 Furthermore, dysregulation of the type I IFN system can elicit autoimmune diseases, perhaps best exemplified by interferonopathies and SLE.5 All 17 type I IFNs signal through the IFN alpha receptor (IFNAR) and induce a type I IFN gene signature (IFNGS), which is present in a proportion of patients with SLE and other autoimmune diseases such as myositis, Sjögren’s syndrome, systemic sclerosis and rheumatoid arthritis.6 7 Recent evidence also implicates type I IFN-dependent signalling as a key inflammatory driver in non-autoimmune diseases such as certain solid tumours and myocardial infarction.8 9
The second International Summit on Interferons in Inflammatory Diseases, sponsored by AstraZeneca, was held in Gaithersburg, Maryland, USA (17–18 May 2018) and united 26 international expert clinicians and scientists with diverse backgrounds in basic science, translational science and clinical medicine. In this review, we build on the content of the inaugural meeting10 by reviewing high-impact research on the role of type I IFNs in autoimmune and other inflammatory diseases published in recent years.
Overview of the type I IFN system in host defence
The primary (or ‘professional’) IFN-producing cells in antiviral innate immunity are plasmacytoid dendritic cells (pDC).11 Type I IFN expression is triggered by nucleic acid-sensing pattern recognition receptors, such as endosomal membrane-bound toll-like receptors (TLR; DNA and RNA sensors),12 the cytosolic retinoic acid-inducible gene 1 (RIG-I)-like family of receptors (RNA sensors)13 and cyclic GMP-AMP synthase (cGAS; DNA sensors).14 Type I IFNs induce an IFNGS via IFNAR-dependent activation of Janus kinase (JAK)-signal transducer and activator of transcription (STAT).15
Although they signal through the same receptor, type I IFN subtypes (eg, IFN-α and IFN-β) can have both separate and overlapping roles in host defence.16 Their role in a particular context is determined by differences in timing, signal magnitude and source of the type I IFN subtype. For example, cGAS stimulation preferentially elicits an IFN-β response, whereas TLR stimulation predominately increases IFN-α expression.17 Type I IFNs have pleiotropic effects, which include promoting the maturation of dendritic cells into antigen-presenting cells18 and B cell and T cell survival, activation and differentiation,19 20 in turn promoting further IFN production from pDCs.21 22 Type I IFNs also induce differentiation of B cells into a distinct proinflammatory subset of plasma cells that secrete ISG15.23 Dysregulation of these self-amplifying loops is a hallmark of SLE and other IFN-driven diseases.
Type I IFNs in autoimmune diseases
SLE and interferonopathies
SLE is a chronic inflammatory autoimmune disease that affects many organ systems.24 Although rare forms of monogenic lupus occur, more often a variety of environmental factors trigger SLE in genetically predisposed individuals. It is well established that dysregulation in the type I IFN system is a key driver in SLE pathogenesis and that the interaction of type I IFN with immune cells can induce multifaceted aspects of lupus (table 1).5 6 25–27 Up to 87% of paediatric and adult patients with SLE have an IFNGS.28–31 It remains unclear how the signature relates to the phenotype and disease progression at the cohort and/or individual patient level in established SLE. However, an IFN score was recently shown to predict the development of SLE in at-risk individuals.32 Alternative biomarkers of type I IFN activity have been evaluated and include the IFN-α response protein, sialic acid-binding Ig-like lectin 1, which is expressed exclusively on tissue-resident monocyte-derived dendritic cells and tissue-resident macrophages.33 Other gene signatures, such as plasmablast and neutrophil signatures, may correlate better with SLE disease activity than the IFNGS at the individual level.31
Cellular effects of type I IFN that may contribute to the pathogenesis of lupus
The primary source of type I IFNs in patients with lupus is most likely pDCs, though a role for ‘nonprofessional’ IFN-producing cells (essentially all other nucleated cells, including macrophages) cannot be ruled out. The stimulus for type I IFN production in these patients has not been resolved but may include extracellular and intracellular accumulation of endogenous nucleic acids via increased production (eg, extensive cell damage, apoptosis and NETosis) and/or impaired clearance.34 35 Monocyte-derived macrophages transfected with a small non-coding Y RNA or stimulated with immune complexes were shown to produce IFN-α and IFN-β mRNA transcripts.36 Furthermore, tumour necrosis factor (TNF) can induce macrophages to produce modest amounts of type I IFN, thereby triggering expression of IFN-stimulated genes and a proinflammatory autocrine loop,37 and prolonged exposure to type I IFNs may prime monocytes from patients with SLE to produce a strong inflammasome response to TLR activation.38 This hyperactivity would predispose a patient with SLE to elicit an exaggerated inflammatory response to a subsequent viral infection.
Transposable elements represent nearly half of the human genome and are a significant source of potentially stimulatory self-nucleic acids. In SLE, the expression of transposable elements, such as long interspersed nuclear element type 1 (L1), is dysregulated in tissue and cell-specific patterns and can trigger TLR or cytosolic receptor-dependent type I IFN production.39–42 Overexpression of transposable element RNA may be related to impaired heat shock protein 90 expression in patients with SLE.39
Both endosomal and cytosolic nucleic acid sensors have been implicated in the pathogenesis of SLE; however, it remains unknown if overactivation of either sensor type alone is sufficient to drive disease activity. In neonatal mice, infection with the RNA virus, lymphocytic choriomeningitis virus (LCMV), induces lupus-like disease by 2–5 months of age via both endosomal TLR and cytosolic mitochondrial antiviral signalling (MAVS) protein-dependent type I IFN production.43 LCMV-induced lupus was pDC and endosomal TLR dependent; MAVS signalling alone was insufficient to induce lupus-like symptoms. Recent evidence suggests that exogenous (eg, viral) or endogenous cytosolic RNA may stimulate the DNA sensor, cGAS, by inducing mitochondrial DNA release, cGAS activation and IFN-β production.44 45
Type I IFN and anti-nucleic acid antibodies may collectively set preconditions for altered handling of damaged DNA. Neutrophils primed with type I IFNs and exposed to TLR-activating autoantibodies retain and extrude oxidised mitochondrial DNA, a potent inducer of type I IFN production by pDCs.46 47 Spontaneous activation of MAVS protein in lymphocytes from patients with SLE correlates with mitochondrial oxidative stress and serum type I IFN levels.48 Delineating the nucleic acid triggers in SLE and interferonopathies may inform on novel drug targets. For example, pathway elements that potential therapies could target have been identified in an in vitro model of Aicardi-Goutières syndrome, including the activation of cGAS-dependent type I IFN production by increased cytosolic reverse-transcribed DNA (figure 1A).49 Thus, reverse transcriptase inhibition may be a potential strategy to treat patients with this disease.
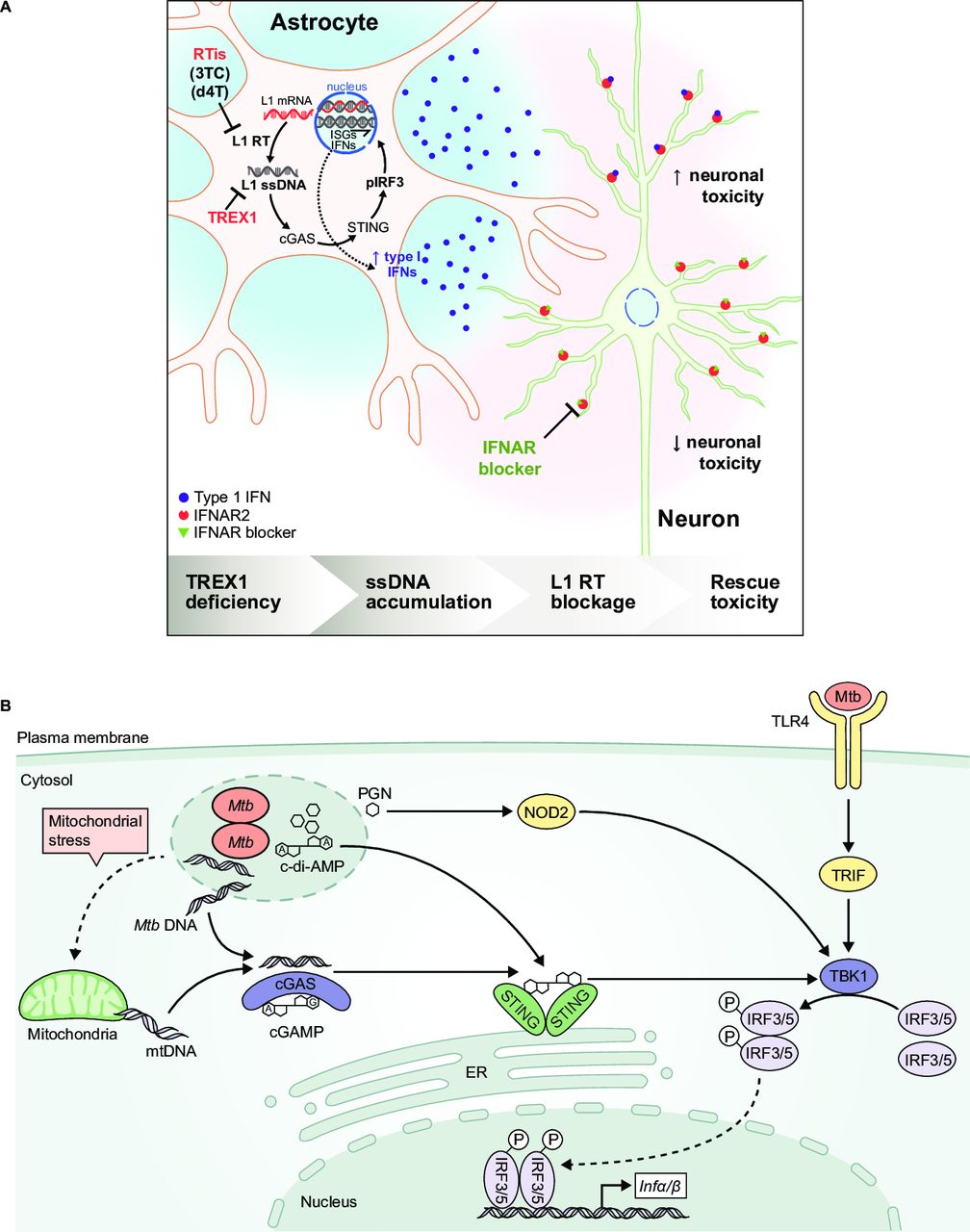
{kind=link}
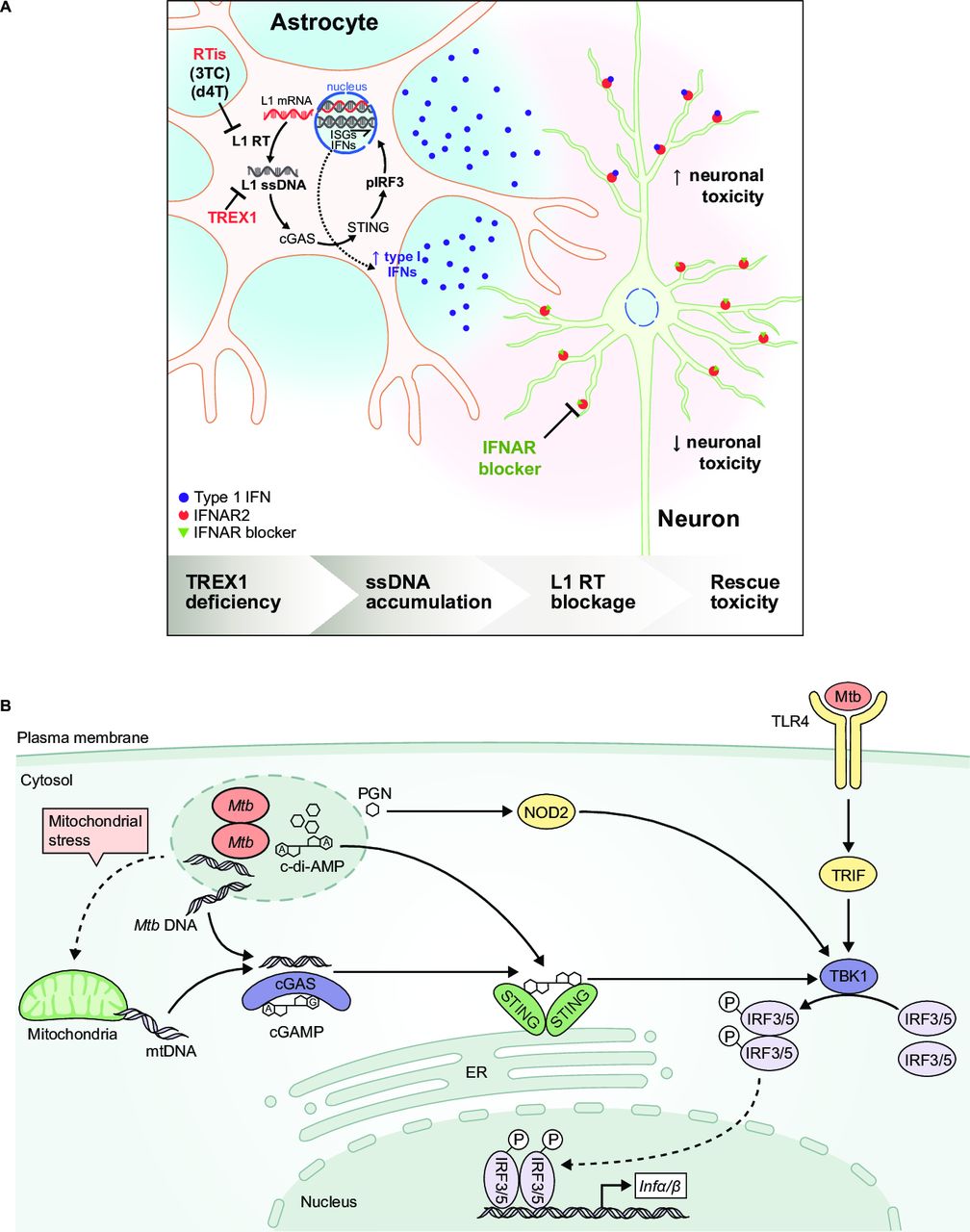
Comparison of nucleic acid signalling pathways leading to type I interferon (IFN) production in an autoimmune disease and bacterial infection. (A) In vitro model of Aicardi-Goutières syndrome. Mutations in antiviral genes, including three-prime repair exonuclease 1 (TREX1), can cause Aicardi-Goutières syndrome, an autosomal recessive progressive inflammatory disorder. TREX1-deficient human neurons accumulate long interspersed element-1 (L1) ssDNA, which is produced by the reverse transcription of L1 retrotransposon transcripts. L1 ssDNA stimulates the cGAS-STING pathway, resulting in the production of neurotoxic IFN. Neurotoxicity could be blocked by reverse transcriptase inhibitors (RTis) and IFN-α receptor (IFNAR) blockers.49 Reprinted with permission from Elsevier and Thomas CA, et al.49 (B) Type I IFN induction through alternative pathways during Mycobacterium tuberculosis infection. Mycobacterial (Mtb) infection results in the production of microbial products and products associated with mitochondrial stress that stimulate pattern recognition receptors, including TLR4, NOD2 and cGAS, to activate type I IFN gene transcription (adapted from Moreira-Teixeira et al [96]). cGAS, cyclic GMP-AMP synthase; IFN, interferon; IFNAR, IFN alpha receptor; IRF, IFN regulatory factor; STING, stimulator of IFN gene; TLR, toll-like receptor.
Recent reports provide insight into the genetic background that increases type I IFN expression and contributes to SLE risk. A global transancestral association study of SLE using genomic data from more than 27 000 individuals, including 11 590 patients with SLE, confirmed that SLE risk has both ancestry-dependent and ancestry-independent contributors.50 Hagberg et al demonstrated increased T cell STAT4 responsiveness to interleukin (IL)-12 and IFN-α in patients with SLE carrying the STAT4 risk allele.51 Furthermore, healthy individuals with the risk gene variant have normal STAT4 responsiveness to IL-12, which can become ‘lupus like’ if cells from these individuals are exposed to IFN-α.52 The presence of the purine nucleoside phosphorylase risk allele was associated with increased type I IFN-induced mRNA expression in B cells derived from patients with SLE.53
Epigenetic mechanisms alter gene expression and contribute to SLE heterogeneity.54 DNA methylation profiles were analysed in an epigenome-wide association study of more than 500 patients with SLE and a similar number of controls.55 Differential methylation of type I IFN-regulated genes was most notable for patients with active versus inactive disease. In SLE-discordant twins, differential methylation was present in type I IFN-regulated genes for T and B cells, monocytes and granulocytes, and hypomethylation of these genes was associated with increased SLE flare risk.56 Park et al57 employed a comprehensive epigenomics approach to examine cross-regulation of TLR responses at the level of chromatin in macrophages by TNF and type I IFNs.57 TNF-induced silencing of TLR signalling was prevented by type I IFN-induced priming of chromatin. These data may explain why some patients with chronic inflammatory diseases become severely ill when they develop a subsequent infection.
Sjögren’s syndrome
Sjögren’s syndrome is an autoimmune disease that primarily affects the exocrine glands, with strong evidence that dysregulation of the type I IFN system is a key driver of inflammation.58 59 However, the effects of type I IFNs in Sjögren’s syndrome may be subtype specific. IFN-β may reduce the expression of proinflammatory mediators in peripheral blood mononuclear cells isolated from patients with Sjögren’s syndrome.60 Like SLE, patients with Sjögren’s syndrome can be stratified by those who do and do not have an IFNGS. Patients with the signature have increased B cell activating factor (BAFF) expression,61 which is involved in B cell activation, and a higher prevalence of autoantibodies to Sjögren’s syndrome-related antigen A (SSA; also called anti-Ro/SSA) and B (SSB; also called anti-La/SSB) than those without the signature.62 Patient stratification also may be possible based on the presence or absence of a type II IFNGS.63 In a phase 2 study, to evaluate the effects of belimumab, an anti-BAFF antibody, on exocrine inflammation in patients with Sjögren’s syndrome,64 a low blood and salivary count of natural killer cells was the only predictor of response to belimumab. The authors proposed that two subpopulations of patients with Sjögren’s syndrome may exist: one with a predominant type I IFN-BAFF-B cell axis (ie, belimumab responders) and another with a predominant type II IFN axis associated with natural killer cell activity.
Systemic sclerosis
Systemic sclerosis is an atypical autoimmune disease in that both inflammatory (ie, vasculopathy) and non-inflammatory (ie, dermal and visceral fibrosis) processes contribute to clinical manifestations.65 Dermal pDCs and a dysregulated type I IFN system are implicated in the clinical manifestations of systemic sclerosis, including fibrosis,66–70 and an IFNGS is present in more than 68% of patients.28 pDCs in the skin of patients with systemic sclerosis aberrantly express TLR8, which is responsible for pDC secretion of chemokine (C-X-C motif) ligand 4 (CXCL4; also called platelet factor 4), and TLR8 and TLR9-induced type I IFN production by pDCs is potentiated by CXCL4.70 Aberrant TLR8 expression and subsequent secretion of both CXCL4 and IFN-α by pDCs may partially explain why two IFN-driven diseases, lupus and systemic sclerosis, can have such distinct clinical manifestations. Targeting pDCs rather than a specific IFN may be a more effective approach to treating patients with systemic sclerosis.
Myositis
Idiopathic inflammatory myopathies are heterogeneous, systemic autoimmune diseases with muscle (and often skin) as the primary target(s) and include polymyositis and dermatomyositis.71 Muscle biopsies from patients with dermatomyositis are characterised by overlapping distributions of large numbers of pDCs, a prominent IFNGS and abundant type I IFN-inducible protein, myxovirus resistance protein 1, underscoring the potential role of type I IFNs in driving disease activity.72 Furthermore, the IFNGS and levels of type I IFN-regulated chemokines in blood correlate with disease activity in patients with dermatomyositis. IFN-β is the predominant type I IFN subtype in the sera of patients with dermatomyositis and correlates with the IFNGS.73 Consistent with these findings, severe dermatomyositis can be triggered by IFN-β therapy for multiple sclerosis (MS).74 Although IFN-β predominates, IFN-α has also been implicated in the pathogenesis of dermatomyositis. Piper et al75 have shown that IFN-α drives the expansion of an immature transitional B cell population with a proinflammatory phenotype in juvenile dermatomyositis.75
Rheumatoid arthritis
Rheumatoid arthritis is a chronic inflammatory autoimmune disease that primarily affects the joints, but as a systemic disease it has extra-articular manifestations in the eyes, heart, lungs and other organs.76 pDCs and IFN-α/β levels are increased in the rheumatoid arthritis synovium compared with the joints of healthy individuals,77–79 and up to 50% of patients with rheumatoid arthritis have a peripheral blood IFNGS.28 80 Baseline type I IFN activity (quantified as type I IFN protein and IFNGS expression) may predict clinical responders to TNF antagonists and non-responders to rituximab in patients with rheumatoid arthritis.81–83 However, the IFNGS may not reflect rheumatoid arthritis disease activity,84 and whether there is a causal relationship between IFNGS and rheumatoid arthritis pathogenesis is currently unclear. Indeed, recent evidence suggests that pDCs from drug-naïve patients with early rheumatoid arthritis differentially expressed genes suggestive of enhanced tolerogenic function.85
Autoimmune regulator-deficient patients
Autoimmune regulator (AIRE) is a transcriptional regulator that promotes clonal depletion of self-reactive T cells. Highlighting this role, AIRE deficiency causes autoimmune polyglandular syndrome type 1 (APS-1; also known as autoimmune polyendocrinopathy candidiasis-ectodermal dystrophy/dysplasia).86 AIRE-deficient individuals have autoreactivity against self-antigens, including those typically associated with MS, SLE, type I diabetes mellitus and rheumatoid arthritis. Paradoxically, it is not clear that MS and SLE have ever been described, and the other two conditions are more rare in AIRE-deficient individuals than might have been expected. Casting light on this, Meyer et al87 performed protoarray analyses and additional techniques to investigate sera from patients with APS-1 and controls.87 88 In addition to global loss of T cell tolerance, patients with APS-1 had two types of B cell dysregulation: (1) diverse or ‘private’ reactivities of up to 100 diverse gene products, many of which were AIRE regulated; and (2) shared reactivities to steroidogenic enzymes and selected cytokines, none of which were obviously AIRE regulated. Remarkably, high-affinity antibodies to IFN-α were present in almost all patients, preventing IFNAR-dependent signalling, and were negatively correlated with the prevalence of type I diabetes mellitus in patients who carried other pathognomonic features, such as antibodies to glutamic acid decarboxylase (GAD)65 and GAD67. Thus, specific autoantibodies may actively limit certain diseases in AIRE-deficient individuals. Interestingly, the presence of anti-IFN-α antibodies did not predispose patients with APS-1 to severe viral infections, which may reflect preserved antiviral protection from IFN-β or other IFNs. The high-affinity anti-IFN antibodies obtained from the blood of patients with APS-1 have been useful in providing tools of unprecedented sensitivity for measuring IFN-α levels in human body fluids.89
The type I IFN system in non-autoimmune diseases
Tuberculosis
Infection with Mycobacterium tuberculosis is the leading cause of mortality from infectious diseases.90 91 In most individuals, M. tuberculosis infection is controlled by the host immune response, with CD4+ T cells, IL-12, IFN-γ and TNF as the most critical factors.92 It remains unclear why some people are not protected from developing active tuberculosis (TB). Patients with active TB can be distinguished from those with latent TB by the presence of a peripheral blood gene signature with increased expression of type I IFN-inducible genes and decreased expression of IFNG and TBX21.93 Furthermore, the type I IFN-induced components of the gene signature correlate with radiographic evidence of active TB and diminish with successful treatment.94 95 These findings and the body of evidence from mouse models of TB support a role for type I IFNs in the pathogenesis of TB.96
Distinct strains of M. tuberculosis variably induce type I IFN. Recognition of one particular strain by TLR4 was associated with production of IFN-β and increased virulence, with lung pathology observed early in the course of infection.97 Specific M. tuberculosis strains can differ in their capacity to induce mitochondrial stress, generation of reactive oxygen species and release of host mitochondrial DNA into the cytosol.98 The release of mitochondrial DNA contributes to cGAS and stimulator of IFN gene (STING)-dependent production of IFN-β after M. tuberculosis infection98–101 (figure 1B).
Type I IFN effects on TB are context specific. In mouse models of TB, type I IFNs induce the production of the immunosuppressive cytokine IL-10, reduce production of protective cytokines such as IL-1 and impair the macrophage response to type II IFN (IFN-γ). This contributes to decreased induction of Th1 adaptive immunity, increased bacterial loads and shorter survival times.102–107 In contrast to data from the murine models, several clinical studies have reported beneficial effects of type I IFN administration in the setting of well-established M. tuberculosis infections.108 109 Additionally, in a low IFN-γ state or in the setting of reduced IFN-γ signalling, low levels of type I IFN may maintain the function of classically activated protective macrophages by inhibiting Arg1 expression and the related conversion of protective macrophages to an alternatively activated, less protective phenotype.110 111
Understanding the mechanisms responsible for switching from acute to sustained type I IFN expression in chronic M. tuberculosis infection may be informative for autoimmune diseases, such as SLE, in which activation of latent Epstein-Barr virus has been suggested as a factor that can trigger autoimmunity in a genetically predisposed individual.112 113
HIV infection
HIV infection results in profound immune system dysfunction, classically characterised by progressive CD4+ T cell depletion.114 Despite the availability of improved treatments, many patients experience chronic inflammation associated with sustained production of type I IFN and its sequelae.114 The effects of type I IFN in the setting of HIV-1-induced inflammation may be IFN subtype specific. In the HIV-exposed brain, IFN-α promotes neuropathology, whereas IFN-β is neuroprotective.115 A relationship may exist between the sustained IFNGS and disease progression. Simian immunodeficiency virus (SIV) infection is non-progressive in African green monkeys and is associated with a transient IFNGS after acute infection. In contrast, SIV infection in Asian macaques is progressive and associated with a sustained IFNGS.116 It remains unclear if this relationship is correlative or causative.
HIV-1 pathogenesis, including the role of pDCs and type I IFNs, has been studied in humanised mice, which are immunodeficient animals stably reconstituted with human immune cells/tissues to provide an in vivo functional human immune system that is tolerant to both human and mouse antigens.117 In this model, the pDC/type I IFN axis has distinct roles in acute and chronic HIV infection. HIV enters pDCs through CD4-dependent endocytosis and induces type I IFN production primarily through RNA-mediated activation of TLR7.118 In acute HIV infection, pDCs contribute to suppression of HIV replication and promote priming of anti-HIV-1 T cells.119 In chronic HIV infection, sustained type I IFN production from pDCs contributes to depleting/exhausting T cells.119–121 Thus, pDCs can have opposing roles depending on the stage of HIV infection.
During treatment with antiretroviral agents, HIV replication is suppressed and viral antigen production decreases below detectable levels in the blood and other tissues. Cells with HIV-1 DNA persist in tissue, and, if antiviral treatment ceases, viral replication rebounds within 2 weeks. The dependence on combination antiretroviral therapy (cART) for viral replication suppression can be replicated in the humanised mouse models. These models have been used to determine if targeting the pDC/type I IFN axis in the treatment of patients with organ inflammation resulting from chronic HIV infection could also prevent viral rebound on cART withdrawal. IFNAR blockade in HIV-1-infected humanised mice fully reversed HIV-1-induced immune hyperactivation, rescued anti-HIV-1 immune responses in T cells, decreased HIV-1 reservoir size and delayed rebound after stopping cART.121 Theoretically, an anti-type I IFN approach to treating chronic HIV would not require lifelong treatment because the goal is to rescue the T cell population, augmenting elimination or control of the virus. If recovery of T cell function can be sustained, future studies might assess whether anti-type I IFN therapy diminishes viral reservoirs and eliminates viral replication, a situation that might be considered a ‘functional cure’.
Oncology
Antigenicity, adjuvanticity and homeostatic feedback are major discriminatory functions of the immune system that are critical in cancer biology and regulated, in part, by the type I IFN system.122 Low levels of type I IFN in the tumour have an anticancerous effect by activating T cell-dependent adaptive immunity, whereas higher levels are effective by inhibiting angiogenesis.123 Conventional cancer therapies, such as radiation, chemotherapy and epigenetic drugs, can activate the type I IFN system and stimulate the immune response to cancer.122 124 Genotoxic cancer therapies lead to breaks in genomic DNA, which in turn can act as stimuli for the pattern recognition receptor, cGAS, and induce IFN signalling.124
Recent evidence suggests that type I IFN signalling also may have a detrimental role in the immune tumour microenvironment. Patients with IFNGS-positive breast cancer tumours are more likely to fail chemotherapy than patients with IFNGS-negative tumours.125 In its unshielded state, RN7SL1A, a prominent stromal fibroblast RNA, can elicit a RIG-I-dependent IFNGS.126 127 Carcinoma-associated basement membrane disruptions promote fibroblast production of exosomes containing unshielded RN7SL1A, which in turn transfer the unshielded RNA to neighbouring breast cancer cells.126 127 In breast cancer cells, RIG-I-dependent activation of STAT1 elicits a NOTCH3-dependent pathway that can expand therapy resistance, progression and metastasis in breast cancer.127 In another study, relapse after radiation therapy and anti-cytotoxic T-lymphocyte-associated protein 4 treatment was associated with prolonged type I IFN signalling in mice.128 In summary, IFN can have a positive or negative influence on cancer growth, and this influence may depend on various factors including treatment, cancer type, tumour microenvironment and level of IFN stimulation.
Myocardial infarction
Coronary atherosclerosis develops asymptomatically until acute myocardial infarction elicits chest pain, forcing patients to seek medical care.129 In the first few days after infarct, cardiomyocyte cell death is followed by a sterile inflammatory response that eventually resolves into fibrosis.129 130 Understanding the mechanisms of pathological ventricular remodelling is an area of intense research effort.
On myocardial ischaemia, with events including double-stranded DNA breaks,131 damage-associated molecular patterns (DAMPs) may be liberated after the ischaemia event and may contribute to the inflammatory response by inducing type I IFN. In a mouse model of myocardial ischaemia, an IFN regulatory factor 3 (IRF3)-dependent IFNGS was upregulated in cardiac macrophages 4 days after infarction.9 The IFNGS required cGAS and IFNAR, implicating endogenous nucleic acids as the trigger for a type I IFN response. Self-DNA was released by injured cardiomyocytes and taken up by macrophages. IRF3 or IFNAR-deficient mice had improved survival rates after the injury. Administration of an anti-IFNAR antibody within 48 hours after myocardial ischaemia decreased the inflammatory response, reduced ventricular dilation and improved cardiac performance compared with mice that did not receive treatment. Thus, anti-type I IFN drugs may be beneficial in the acute postmyocardial ischaemia period.
Conclusions
SLE remains the prototypic type I IFN-driven disease, and evidence is accumulating for targeting the type I IFN pathway as a rational therapeutic approach. In recent years, there has been considerable effort to develop drugs for SLE and interferonopathies, and clinical trial data support the strategy of targeting signalling components downstream of type I IFNs for the treatment of type I IFN-driven diseases. Anifrolumab, a fully human monoclonal antibody against the IFNAR, plus standard of care neutralised the IFNGS, decreased SLE disease activity relative to placebo and was well tolerated in a phase 2b trial with patients with SLE.29 Baricitinib, a JAK inhibitor, decreased SLE disease activity in patients with SLE in a phase 2 trial132 and improved symptoms, decreased corticosteroid dosage and neutralised the IFNGS in patients with other type I interferonopathies, including chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures and STING-associated vasculopathy with onset in infancy.133
The presence of an IFNGS may predict clinical responders to treatment, but patients without a strong IFNGS remain an important subpopulation with clinically significant disease activity. To date, serious treatment-related adverse events have not been observed in patients receiving anti-type I IFN-targeted therapy, which is intriguing because the type I IFN system is fundamental to both innate and acquired immunity.29 132 pDC depletion may be a more selective approach to controlling excessive type I IFN production in autoimmune diseases because the ‘nonprofessional’ IFN-producing cell populations would be spared. At the same time, the community will benefit in the near future from a better understanding of the heterogeneity of pDCs. BIIB059, a humanised monoclonal antibody to blood dendritic cell antigen 2 (a pDC-specific receptor), is currently being developed for the treatment of SLE. In a phase 1 study, it demonstrated a favourable safety profile, decreased IFN-response gene expression and reduced skin disease activity.134
Substantially more research is needed to discern the mechanisms of induction and response to type I IFNs across autoimmune and non-autoimmune inflammatory diseases, especially with regards to identifying the drivers of type I IFN production and the effects of these cytokines on T cell populations. Sustained type I IFN production in chronic infections contrasts with the transient and well-controlled response after acute viral infections but may be mechanistically similar to the dysregulation observed in the type I IFN system for IFN-driven autoimmune diseases.
The role of type I IFNs in cancer and chronic infections is complex and context specific—at times they are ‘friend’ and at other times they are ‘foe’—but this does not preclude therapeutically targeting the type I IFN pathway at an appropriate stage of the disease process. The potential efficacy of targeting the type I IFN pathway in humans is evident from the correlates of anti-IFN antibodies and clinical symptoms in subcohorts of patients with APS-1. Patient phenotyping will be critical to successful intervention and will be helped by the bourgeoning capacity to accurately measure type I IFN-α. Given the prevalence of cancer and chronic TB/HIV infections, identifying drug targets along the type I IFN system for these therapeutic areas has the potential to yield treatments that would greatly impact human health around the world.
Supplemental material
Acknowledgments
The second International Summit on Interferons in Inflammatory Diseases was sponsored by AstraZeneca. Editorial support was provided by Francis Golder, BVSc, PhD, and Alan Saltzman, PhD, of JK Associates. We thank the IFN summit attendees (online supplementary appendix table 1) for their participation and help in reviewing this manuscript.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.
- 136.
- 137.
- 138.
- 139.
- 140.
- 141.
- 142.
- 143.
- 144.
- 145.
- 146.
Footnotes
Funding This study was supported by AstraZeneca.
Competing interests MC and LR, as well as the meeting participants, received fees for their time in preparing for and presenting at/attending the IFN Summit meeting. They received no fees for their work as authors of the manuscript.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; internally peer reviewed.
Data availability statement No additional data are available.