Article Text

Abstract
Objective We compared the physician-assessed diagnostic likelihood of SLE resulting from standard diagnosis laboratory testing (SDLT) to that resulting from multianalyte assay panel (MAP) with cell-bound complement activation products (MAP/CB-CAPs), which reports a two-tiered index test result having 80% sensitivity and 86% specificity for SLE.
Methods Patients (n=145) with a history of positive antinuclear antibody status were evaluated clinically by rheumatologists and randomised to SDLT arm (tests ordered at the discretion of the rheumatologists) or to MAP/CB-CAPs testing arm. The primary endpoint was based on the change in the physician likelihood of SLE on a five-point Likert scale collected before and after testing. Changes in pharmacological treatment based on laboratory results were assessed in both arms. Statistical analysis consisted of Wilcoxon and Fisher’s exact tests.
Results At enrolment, patients randomised to SDLT (n=73, age=48±2 years, 94% females) and MAP/CB-CAPs testing arms (n=72, 50±2 years, 93% females) presented with similar pretest likelihood of SLE (1.42±0.06 vs 1.46±0.06 points, respectively; p=0.68). Post-test likelihood of SLE resulting from randomisation in the MAP/CB-CAPs testing arm was significantly lower than that resulting from randomisation to SDLT arm on review of test results (−0.44±0.10 points vs −0.19±0.07 points) and at the 12-week follow-up visit (−0.61±0.10 points vs −0.31±0.10 points) (p<0.05). Among patients randomised to the MAP/CB-CAPs testing arm, two-tiered positive test results associated significantly with initiation of prednisone (p=0.034).
Conclusion Our data suggest that MAP/CB-CAPs testing has clinical utility in facilitating SLE diagnosis and treatment decisions.
- systemic lupus erythematosus
- clinical utility
- laboratory test
- diagnosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE remains one of the most challenging autoimmune rheumatic diseases to diagnose in rheumatology practice and is a leading cause of death in young females.1 The difficulties in the ability of healthcare providers to make a diagnosis of SLE is primarily related to the low prevalence of the disease and the heterogeneity of symptoms that are often non-specific and overlapping with many other conditions such as fibromyalgia.2 Diagnostic challenges are also due in part to the lack of diagnostic criteria and limitations with current diagnostic immunology testing, primarily antinuclear (ANA) and anti-double stranded (ds) DNA antibodies that lack specificity and sensitivity, respectively.3 It follows that the validation and introduction in clinical practice of novel SLE markers or testing methods that facilitate the diagnosis of the disease is an unmet need for the rheumatologists. Early diagnosis can identify patients likely to benefit from treatment (such as hydroxychloroquine (HCQ)) to limit organ damage and decrease healthcare utilisation.4 In addition, accurate determination of the unlikely SLE diagnosis in ANA-positive patients with non-specific symptoms is important and can have direct benefit by decreasing inappropriate referrals to the rheumatologist.
Complement hyperconsumption due to activation of the complement system is intimately associated with SLE and measurement of serum complement levels is now integrated in modern classification criteria for the disease.5 Over the past two decades, many studies have reported that quantification of cell-bound complement activation products (CB-CAPs) is a valid measure of classical complement pathway activation and has demonstrated value in SLE diagnosis and monitoring.3 6 A multianalyte assay panel (MAP) that combines CB-CAPs (C4d on erythrocytes (EC4d) and B cells (BC4d)) with eight autoantibodies has been developed to assist rheumatologists with the differential diagnosis of SLE6 and multiple studies support the clinical validity and accuracy of the panel in distinguishing SLE from a variety of other rheumatic diseases.3 7–10 Furthermore, MAP and CB-CAPs consistently outperform serum complement levels for SLE diagnosis. A case–control, retrospective review of medical charts also suggested the clinical utility of MAP/CB-CAPs laboratory testing in assisting rheumatologists in real-world practice.11 However, prospective data comparing standard diagnosis laboratory testing (SDLT) to MAP/CB-CAPs laboratory testing is lacking. In this randomised study, our objective was to assess prospectively the clinical utility of diagnostic immunology testing and MAP/CB-CAPs in assisting rheumatologists with the diagnosis of SLE. We also evaluated whether treatment decisions were affected by the treatment arms to which the patients/physicians were assigned.
Methods
The Clinical Laboratory Assessment and Recommendation for Lupus (CARE for Lupus) was a multicentred, randomised and prospective study in the USA that enrolled patients referred to board-certified rheumatologists with a suspicion of SLE. Patient consent was collected for all patients. At enrolment, all patients presented with a history of ANA positivity within the prior 6 months and were assessed clinically within 3 months of their referral to rheumatologists. Patients were randomised to two different groups (1:1) on the day of enrolment using block randomisation of 20 patients. For the SDLT group, no recommendation was made to the rheumatologists with regard of which laboratory tests to order and what laboratory to order them from. The other randomisation group corresponded to the MAP/CB-CAPs laboratory testing method. Both the sponsor and investigators were blinded to the randomisation list for the duration of the study. MAP/CB-CAPs combines CB-CAPs (erythrocyte bound C4d (EC4d) and B-lymphocyte bound C4d (BC4d)) with eight autoantibodies (ANA, anti-Smith, anti-dsDNA (confirmed using Crithidia indirect immunofluorescence (IIF)), anticyclic citrullinated peptide (CCP), anti-centromere B (CENP), anti-Jo1, anti-Scl-70 and anti-SSB antibodies) to produce a two-tiered index value, in which a positive test result (>0 index value combining tier 1 and tier 2) results into 80% sensitivity and 86% specificity in distinguishing SLE from a control group of patients with other autoimmune rheumatic diseases.3 Details about the two-tiered method is provided in the online supplementary figure 1. MAP/CB-CAPs index value was measured at Exagen (Vista, California, USA), which runs a clinical laboratory accredited by the College of American Pathologists as currently approved by the Clinical Laboratory Evaluation Program in the state of New York.6 For all patients, venous blood was collected at the time of clinical assessment and randomisation and was shipped overnight to Exagen in transportation kits. The MAP/CB-CAPs index value was reported to clinicians only for the patients randomised to the MAP/CB-CAPs testing arm. In addition to the index value, autoantibody levels were reported individually and were considered positive or negative based on the manufacturer cutoffs. EC4d and BC4d were also reported individually as net mean fluorescent intensity (MFI) with abnormal levels above the 99th percentile of a normal healthy group (14 net MFI and 60 net MFI, respectively). ANA titre by indirect immunofluorescence (on NOVA View, Inova Diagnostics, San Diego, California, USA) was also reported. The MAP/CB-CAPs index score was measured also from blood of patients randomised to SDLT arm; however, clinicians remained blinded to test results for the duration of the study.
Supplemental material
Marker results determined in the group of patients enrolled in the CARE study were compared with a database of 283 754 specimens submitted to the clinical laboratory for diagnosing SLE. All specimens collected for the study were processed with the daily clinical load and quality system management in place in the clinical laboratory in accordance to standard operating procedures.
A five-point Likert scale (0: very low; 1: low; 2: moderate; 3: high; 4: very high) estimating the physician likelihood for SLE was collected pretest and post-test. Per protocol, all patients presented with a low to moderate likelihood of SLE at enrolment (1 or 2 points on the Likert scale), and all cases were adjudicated by lupus experts (AW and EM) at the time of enrolment. An adjudicator determined whether there was agreement that the patient met the criteria for low or moderate likelihood of SLE based on clinical history, physical findings and any laboratory testing prior to enrolment. In all cases when the first adjudicator disagreed with the investigator and in a subset of cases when the first adjudicator agreed with the investigator, a second adjudication was performed (blinded as to agreement or disagreement). If both adjudicators disagreed with the investigator (too high or too low likelihood of SLE), then the patient was considered a screen failure. Both adjudicators remained blinded as to group assignment and laboratory testing post-enrolment. During follow-up, the post-test likelihood of SLE was also collected using the five-point Likert scale and its change before and after testing (on review of test results and 12 weeks after enrolment) was used to quantify clinical utility. Patient-reported outcomes measuring health-related quality of life using five-level EQ-5D (EQ-5D-5L version) was collected pretesting and post-testing at 12 weeks. Physician global assessment (PGA) of disease activity (0–3 points scale) was collected at enrolment (pretesting) and at 12-week follow-up. Initiation of HCQ or prednisone pretesting and post-testing (on review of test results and 12 weeks after enrolment) was also collected for all subjects. All physician and patient-reported outcomes were collected using web-based interface and electronic data capture (ClinCapture, San Francisco, California, USA). Statistical analysis (using R) consisted of Wilcoxon, Kruskal Wallis, Fisher’s exact and linear mixed-effect models as appropriate.
Results
Patients and laboratory results
A total of 145 subjects were enrolled at 32 sites between July 2017 and December 2018 and randomised to SDLT arm and MAP/CB-CAPs testing arm. Enrolment details are summarised in online supplementary figure 2. At enrolment, pretest likelihood of SLE was similar in the SDLT and MAP/CB-CAPs testing arms (1.42±0.06 vs 1.46±0.06 points, respectively; p=0.68). Patient demographics are summarised in table 1. Signs and symptoms at enrolment in each of the randomisation group are presented in the online supplementary table 1. The components of the MAP/CB-CAPs panel in each group is presented in the online supplementary table 2; positivity rate was comparable across randomisation groups (p>0.50) and consistent with that of a population of patients tested in the clinical laboratory during a 6-year period (2012–2018), except for ANA as determined by IIF (81% vs 60%; p<0.01). The positivity rate for the two-tiered index value was 12.5% in the MAP/CB-CAPs testing arm (2.7% tier 1 positive and 9.8% tier 2 positive) and 15.1% in the SDLT arm (2.8% tier 1 positive and 12.3% tier 2 positive). Diagnostic immunology tests ordered in the group of patients randomised to SDLT arm are presented in online supplementary table 3.
Demographics at enrolment
Diagnosis
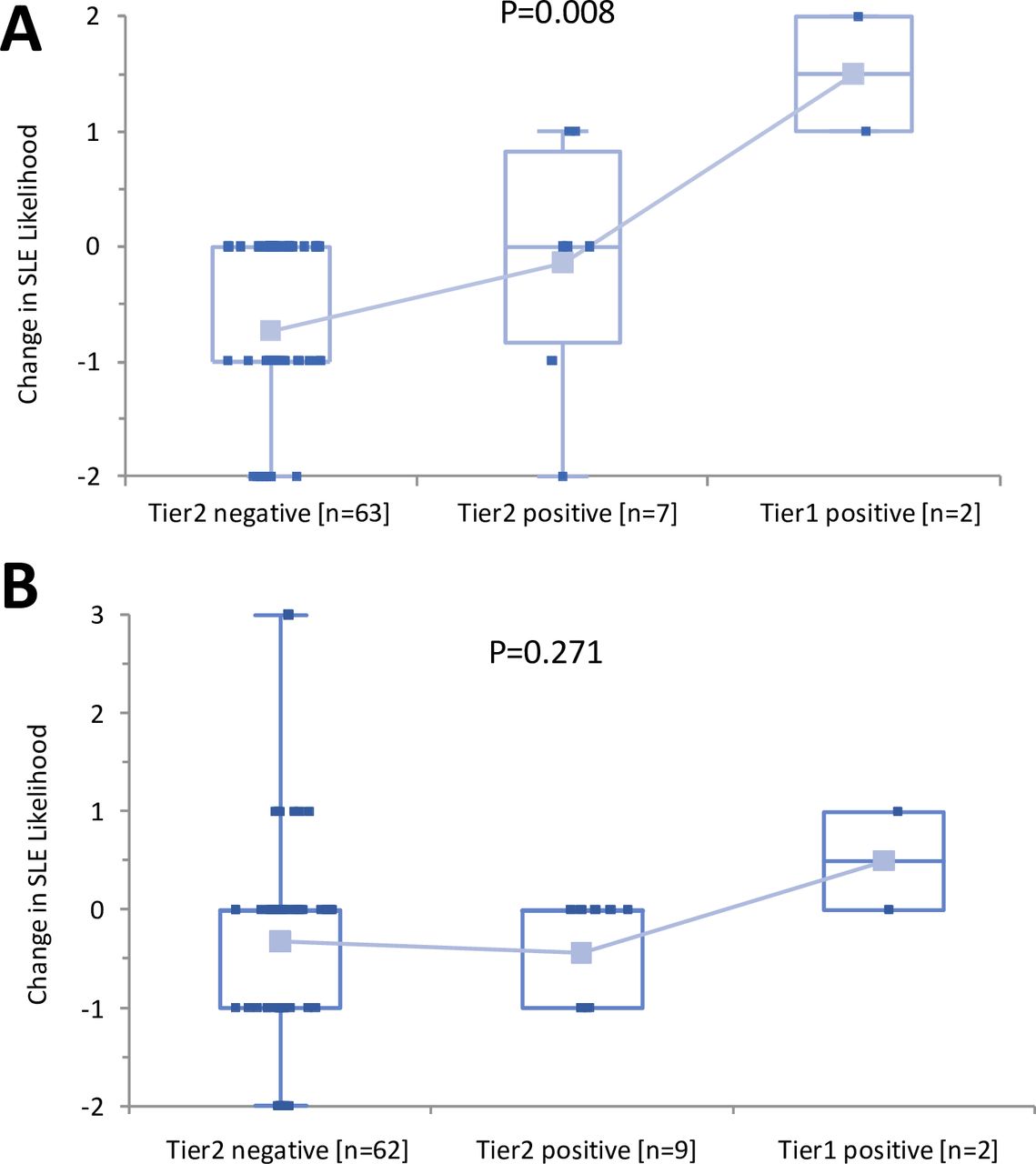
On review of test results, there was a significant decrease in post-test likelihood of SLE, irrespective of randomisation arm (Wilcoxon paired test; p<0.01). Results are presented in table 2. Post-test likelihood of SLE resulting from randomisation to the MAP/CB-CAPs testing arm was lower than that resulting from randomisation to the SDLT arm (−0.44±0.10 points vs −0.19±0.07 points, respectively; p=0.027) and this significant impact on decreasing the likelihood of SLE remained significant at the 12-week follow-up visit (−0.61±0.10 points vs −0.31±0.10 points, respectively; p=0.025). At the 12-week visit, a significant greater decrease in the likelihood of SLE (decrease ≥1 point from enrolment) was observed in the group of patients randomised to MAP/CB-CAPs testing arm (40/72, 56%) when compared with the SDLT arm (27/73, 37%) (difference=19%; p=0.031). Figure 1 highlights the change in likelihood of SLE post-test by two-tiered index score. As expected, in the group of patients randomised to the MAP/CB-CAPs arm, a positive two-tiered test score associated with higher post-test likelihood of SLE (p=0.008), in contrast to the group of patients randomised to the SDLT arm (blinded to MAP/CB-CAPs test results; p=0.271).

Change in SLE likelihood from enrolment to week 12 by two-tiered index test results The change in SLE likelihood pretest and post-test is indicated. (A) Multianalyte assay panel/cell-bound complement activation products (MAP/CB-CAPs) randomisation group; (B) standard diagnosis laboratory testing (SDLT) randomisation group. Kruskal-Wallis analysis of variance p values are provided for each group.
Physician-reported likelihood of SLE pretest and post-test
Overall, among patients negative for MAP/CB-CAPs test results (125 subjects), a lower likelihood of SLE (decrease ≥1 point from enrolment) was detectable at 12 weeks in the group of patients randomised to MAP/CB-CAPs testing arm (38/63, 60%) when compared with the SDLT arm (23/62, 37%) (difference=23%) (p=0.012). None of the patients randomised to the MAP/CB-CAPs testing arm presented with higher post-test likelihood of SLE in the presence of negative test results. Conversely, among patients positive for MAP/CB-CAPs (20 subjects), a higher likelihood of SLE (increase ≥1 point from enrolment) was detectable at 12 weeks (4/9, 44%) in patients randomised to the MAP/CB-CAPs testing arm when compared with the SDLT arm (1/11, 9%) (difference=35%; p=0.127). Results are presented in the online supplementary table 4. Linear mixed-effect models also revealed that the post-test likelihood of SLE decreased in SDLT (estimate: −0.16±0.04 per visit of follow-up; p<0.01) and MAP/CB-CAPs testing arms (estimate: −0.31±0.04 per visit of follow-up; p<0.01). Multivariate linear mixed-effect models established that post-test likelihood of SLE associated with randomisation to MAP/CB-CAPs testing arm and positive MAP/CB-CAPs test result after adjusting for follow-up period (table 3).
Linear mixed-effect models of SLE likelihood in relation to testing arm and MAP/CB-CAPs test result (positive or negative).
Other outcomes and treatment
At enrolment, patients presenting with a positive MAP/CB-CAPs test result had higher PGA than those with negative MAP/CB-CAPs test result (1.44±0.14 (n=20) vs 1.10±0.05 (n=125)) (p=0.030). At the 12-week follow-up, a greater improvement in PGA was observed in the MAP/CB-CAPs testing arm (−0.39±0.08 (n=72)) when compared with the SDLT testing arm (−0.29±0.06 (n=73)) but the difference was not significant (p=0.39). Prednisone was started in 10% (7/73) patients randomised to SDLT arm versus 6% in patients randomised to the MAP/CB-CAPs (4/72) (p=0.53). HCQ was started in 25% (18/73) patients randomised to SDLT arm versus 14% (10/72) patients randomised to MAP/CB-CAPs testing arm (p=0.14). The impact of the laboratory testing on the initiation of prednisone and HCQ in the group of patients randomised to the MAP/CB-CAPs testing arm is presented in figure 2 and revealed that the MAP/CB-CAPs test results associated significantly with initiation of prednisone (p=0.034) and a similar trend was observed with initiation of HCQ (p=0.112). The initiation of HCQ or prednisone by marker in the group of patients randomised in SDLT arm is presented in online supplementary table 5 and did not reach significance for any markers (p>0.13).

{kind=link}
{kind=link}
Initiation of prednisone and HCQ by two-tiered test results in MAP/CB-CAPs testing arm. Fisher’s exact p values comparing the three groups are are provided. Prednisone and HCQ were initiated in 22% and 33% patients testing positive for MAP/CB-CAPs (positive tier 1 or positive tier 2), respectively. CB-CAPs, cell-bound complement activation products; HCQ, hydroxychloroquine.
Finally, in the group of patients randomised to the MAP/CB-CAPs testing arm, a positive MAP/CB-CAPs test result associated with increased EQ5D-5L index score from enrolment to visit 2 (estimate: +0.054±0.024; p=0.028), and greater improvements were detectable when compared with the group of patients positive for the MAP/CB-CAPs test result and randomised to the SDLT arm (mean+0.099±0.046 (n=8) vs −0.008±0.050 (n=8); p=0.049) (online supplementary table 6).
Discussion
This study is the first prospective multisite randomised study aimed to compare and quantify the clinical utility of different laboratory testing methods in facilitating the diagnosis of SLE. The clinical utility of antibody systems in the assessment of SLE is undisputed. Although these biomarkers are known to lack ideal diagnostic accuracy, rheumatologists have relied on them to facilitate the differential diagnosis of SLE. Therefore, it is recognised that new technologies that improve standard diagnostic laboratory and immunology testing in SLE are needed. However, the improvement in clinical validity and accuracy of novel diagnostic tests (over standard practice) on their own is insufficient to satisfy the scrutiny from stakeholders in the healthcare system, including payors, clinicians and patients.12 New technologies introduced in clinical laboratory practice must demonstrate their value in the real-life setting. We have previously established that MAP with CB-CAPs has clinical utility in rheumatologist practices, by retrospective chart review,11 using physician-reported likelihood of SLE for patients evaluated for signs and symptoms of SLE.
In this study, our objective was to establish the clinical utility of the MAP/CB-CAPs in clinical practice in a prospective, scientific study. There were some challenges associated with the design of the study, especially due to the lack of consensus and heterogeneity regarding the SDLT in patients who are being considered for a diagnosis of SLE. Therefore, we purposely made no recommendation as to what tests to order or what laboratory to order them from in the group of patients randomised to the SDLT arm, to better reflect common clinical practice. Clinical utility was defined probabilistically and semiquantitatively using the change in the likelihood of SLE, assessed using five-point Likert scale collected pretesting and post-testing. All patients enrolled in the study were referred with an history of ANA positivity and were considered to have a low to moderate likelihood of SLE as determined by the enrolling rheumatologists and the adjudicators, based on prior history and clinical and demographic features at presentation. Since the criteria for entry were a positive ANA and a low to moderate likelihood of SLE, it is not surprising that most of the patients enrolled in this study were negative for the MAP/CB-CAPs index score in both arms. This is comparable to the target population of patients tested in our clinical laboratory. As expected, the likelihood of SLE decreased in the group of patients randomised to SDLT arm, and these data are consistent with the low prevalence of SLE in the population of ANA-positive patients referred to the rheumatologist for a suspicion of SLE.
The lower post-test likelihood of SLE in the group of patients randomised to the MAP/CB-CAPs arm compared with the SDLT arm was observed early on and was maintained at 12 weeks. Importantly, in the presence of MAP/CB-CAPs negative test result, we detected a significantly lower likelihood of SLE in patients randomised to the MAP/CB-CAPs testing arm when compared with the SDLT arm, thereby indicating greater confidence that the diagnosis of SLE is unlikely with the MAP/CB-CAPs testing. Conversely, greater likelihood of SLE was detectable in the presence of a positive MAP/CB-CAPs test result. Altogether, these data are consistent with the greater diagnostic accuracy of the MAP/CBCAPs panel and its ability to influence diagnostic decision-making.
We also collected treatment information and our data revealed that positive two-tiered MAP/CB-CAPs test results led to initiation of prednisone and HCQ treatment, thus suggesting that the MAP/CB-CAPs test results in the context of clinical findings and symptoms were actionable and led to changes in physician’s behaviour pharmacological intervention in that group of patients. On clinical grounds, these patients were deemed to have low to moderate likelihood of SLE. Therefore, it is not surprising that a positive test result would increase their estimate of likelihood of SLE and thus initiation of appropriate treatment such as steroids or HCQ. In contrast, in the group of patients randomised to SDLT, no significant association between the markers and initiation of prednisone or HCQ was detected. There was also significant improvement in the quality of life in the group of patients randomised to the MAP/CB-CAPs treatment arm and given a positive test result, and we speculate that the laboratory information provided may have decreased the uncertainty of the diagnosis for the patient.
We acknowledge that there are potential limitations in our study. First, long-term impact on health status and patient outcome was not collected after 12 weeks. Second, the short time frame of the study did not allow us to collect long-term data, including fulfilment of the classification criteria for SLE or the impact of the diagnostic strategy on formal health outcome including healthcare utilisation. However, the improvement in the diagnostic certainty by rheumatologists with either a negative or positive MAP/CB-CAPs compared with SDLT, and the impact of the positive test results on the initiation of therapy and EQ-5D is likely to associate with changes in outcomes. In conclusion, in this first randomised, prospective, multicentre study, our data suggest the clinical utility of a new diagnostic intervention to aid community-based rheumatologists and the challenging patients they manage.
Acknowledgments
We thank Tarun Chandra for statistical support, John Conklin for data management and Lyssa Friedman for her assistance with clinical operations. We acknowledge the investigators who participated in the CARE study: Dr Ara Dikranian, Dr Atul Singhal, Dr David Lazar, Dr Donald Thomas, Dr Elvia Moreta, Dr Evan Leibowitz, Dr Howard Blumstein, Dr Jamie Pachon, Dr Jose Pando, Dr Joy Schechtman, Dr Phillip Kempf, Dr Puja Chitkara, Dr Rajat Dhar, Dr Reshman Khan, Dr Richard Jimenez, Dr Robert Levin, Dr Ronald George, Dr Sanjay Godhwani, Dr Shariar Cohen-Gadol, Dr Steven Kimmel, Dr Thalia Ramanujam, Dr Theresa Lawrence-Ford, Dr Yesenia Santiago-Casas, Dr Max Hamburger and Dr Howard Busch. We also thank the coordinators and the patients for their participation.
Footnotes
Contributors DW, TD, AW, EM, RA, TP and TO designed the study. TD performed the analysis and wrote the first draft of the manuscript. RA and TO managed the clinical protocol. AW and EM designed the adjudication protocol and performed the adjudications. CI managed and supervised laboratory testing and quality. All authors participated to data collection and approved the final manuscript.
Funding This study was funded by Exagen.
Competing interests DW, AK, MH, KL, SN, JA, YS, MF, RK and EM have received research grants/consulting fees from Exagen. TD, RA, TO, TP, and CI are employed by Exagen. AW is consultant to Exagen.
Patient consent for publication Not required.
Ethics approval Protocol was approved by Institutional Review Boards.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.