Article Text

Abstract
Objective To investigate the pharmacodynamics, pharmacokinetics and safety of cenerimod—a potent, oral, selective sphingosine 1-phosphate 1 receptor modulator—in patients with SLE.
Methods This multicentre, double-blind, placebo-controlled study was conducted in two parts. In part A, patients with SLE were randomised 1:1:1:1 to receive oral cenerimod 0.5, 1 or 2 mg, or placebo once daily for 12 weeks. Following an interim safety review of part A, additional patients were randomised 3:1 for part B and received cenerimod 4 mg or placebo once daily for 12 weeks. Endpoints included changes in total lymphocyte count, SLE Disease Activity Index-2000 (SLEDAI-2K) score (modified (mSLEDAI-2K) to exclude leucopenia), biomarker anti-double-stranded DNA (anti-dsDNA) antibodies, pharmacokinetic assessments and treatment-emergent adverse events (TEAEs).
Results Part A included 49 patients (1:1:1:1 receiving cenerimod 0.5, 1 or 2 mg, or placebo) and part B included 18 patients (13 cenerimod; 5 placebo). Cenerimod caused a statistically significant dose-dependent reduction in total lymphocyte count from baseline to end of treatment (EOT). Compared with placebo at EOT, cenerimod 4 mg had an estimated treatment effect on change from baseline in mSLEDAI-2K score of −2.420 (p=0.0306), and on anti-dsDNA antibodies of −64.55 U/mL (p=0.0082), suggesting clinical and biological improvement in these exploratory efficacy analyses. Trough plasma concentrations were dose proportional and reached steady-state conditions after 4 weeks of once daily dosing. All groups reported similar, non-dose-related frequencies of TEAEs (cenerimod 0.5 mg: 41.7%; 1 mg: 41.7%; 2 mg: 46.2%; 4 mg: 38.5% and placebo: 58.8%). A small, dose-related, non-clinically relevant decrease in heart rate was only observed in the first 6 hours after initiation.
Conclusions With an acceptable safety profile, the efficacy findings suggest that cenerimod has the potential to treat patients with SLE. Further investigation in larger patient populations with longer treatment duration is warranted.
- systemic lupus erythematosus
- cenerimod
- phase II
- sphingosine-1-phosphate receptor
- S1P1
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE is an autoimmune disease that causes multiorgan inflammation.1 Incidence rates for SLE vary greatly worldwide, ranging from around 23 per 100 000 person-years in North America to 0.3 cases per 100 000 person-years in Ukraine.2 SLE is universally more common in women than in men for every age and ethnic group, predominantly affecting women of childbearing age.3 Symptoms directly impact quality of life and can be severely disabling; patients consistently report lower scores on quality-of-life measures than do the general population.4–6 Existing SLE treatments often have serious side effects, especially with long-term use, and contribute to morbidity and mortality.7–10 Therefore, new therapeutic options are needed.
Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid ligand that specifically binds to and activates five known G protein-coupled receptors, S1P1-5, to regulate different physiological and pathophysiological processes.11 Aberrantly activated T and B lymphocytes and the production of autoantibodies play a major pathophysiological role in SLE.1, 12–14 S1P is involved in the egress of lymphocytes from secondary lymphoid organs into the vascular circulation, via the S1P1 receptor, which is highly expressed in endothelial cells and lymphocytes.15 S1P1 receptor modulators block the movement of lymphocytes from lymphoid organs, preventing them from migrating to sites of inflammation.16 Consequently, S1P receptors have become pharmacological targets for autoimmune and inflammatory diseases.17 The therapeutic potential of S1P receptor modulators has been demonstrated in multiple sclerosis with fingolimod, a non-selective S1P receptor modulator, and with siponimod, a selective S1P1,5 receptor modulator; however, S1P1 receptor modulators are not yet available for SLE.18
Cenerimod is a potent, orally active, selective S1P1 receptor modulator with unique signalling properties.19 In the non-clinical setting, cenerimod did not induce bronchoconstriction or vasoconstriction, which are known adverse effects of S1P receptor modulators.19 A phase I study in healthy participants showed that cenerimod was well tolerated with no significant safety concerns across a range of doses from 0.5 to 4 mg once daily.20 The present proof-of-concept study investigated the pharmacodynamics (PD), pharmacokinetics (PK) and safety of cenerimod, and its effect on clinical and biological markers of disease activity in patients with SLE.
Methods
Study design and dosing
The study protocol was approved by the relevant health authority in each country and by an institutional review board or an independent ethics committee at each site. Signed informed consent was obtained from each patient. The study was carried out in accordance with the principles of the International Council for Harmonisation Guidelines for Good Clinical Practice, the Declaration of Helsinki and all applicable national and local laws. This study is registered on ClinicalTrials.gov (NCT02472795).
This multicentre, double-blind, randomised, placebo-controlled 12-week study was conducted at 18 centres across Belarus, Bulgaria, Georgia, Russia, Ukraine and the USA. The study had two parts, part A and part B, which had the same study design: a 30-day screening period followed by a 12-week treatment period, a 6-week follow-up visit, and two telephone calls at 11 and 16 weeks after treatment discontinuation.
In part A, eligible patients were randomly assigned (1:1:1:1) to once daily oral administration of cenerimod 0.5, 1, 2 mg or placebo. After all patients had completed 4 weeks of treatment during part A, an Independent Data Monitoring Committee reviewed non-blinded data in an interim analysis to evaluate the safety profile of cenerimod and recommend whether the study could proceed to part B as planned (study design: online supplementary file 1). In part B, additional patients were randomised (3:1) to once daily oral administration of cenerimod 4 mg or placebo.
Supplemental material
Randomisation was done using an interactive response technology system. The investigator, study site personnel, patients and sponsor personnel involved in the conduct of the study remained blinded to both the treatment allocation and to the interim analysis results until study closure.
Patients
Patients were eligible for inclusion in the study if they met the following criteria: were aged 18–65 years; fulfilled at least four of the American College of Rheumatology revised diagnostic criteria for SLE;21 were diagnosed at least 6 months before screening; had an SLE Disease Activity Index-2000 (SLEDAI-2K) score of at least two points for musculoskeletal or mucocutaneous manifestations; a history of, or positive serum test at screening for, antinuclear antibodies or anti-double-stranded DNA (dsDNA) antibodies; and were receiving background SLE medication (non-steroidal anti-inflammatory drugs, corticosteroids, antimalarials, mycophenolate mofetil, azathioprine or methotrexate) at stable doses for at least 30 days before randomisation. Patients were ineligible if they had severe lupus disease activity (SLEDAI-2K score >12 points), active lupus nephritis, central nervous system lupus or lupus vasculitis within 90 days prior to randomisation. Additional inclusion and exclusion criteria are described in online supplementary file 2.
Procedures
During the study, assessments were conducted at seven scheduled visits (baseline; day 1; weeks 2, 4 and 8 of treatment; at end of treatment (EOT; week 12) and at end of study (EOS; 6 weeks after EOT)). Additionally, patients were contacted via telephone 11 and 16 weeks after EOT to collect safety information, including serious adverse events (SAEs) and pregnancy status.
Assessments at each visit included blood sampling for haematology, clinical chemistry and biomarker analyses; safety and tolerability assessments (monitoring adverse events (AEs; coded using the Medical Dictionary for Regulatory Activities, version 19.0), vital signs, 12-lead electrocardiograms (ECGs), ophthalmic examinations (apart from day 1) and spirometry (apart from day 1) for forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC)); and disease activity assessments (apart from week 2) using the modified SLEDAI-2K (mSLEDAI-2K) to exclude leucopenia because of the mode of action of cenerimod.
During the treatment period, blood samples were collected before dosing at weeks 2, 4, 8 and 12, and at EOS. Trough plasma concentrations (Ctrough) of cenerimod were determined using a validated liquid chromatography coupled to tandem mass spectrometry assay with a lower limit of quantification of 0.1 ng/mL.
Predefined day 1 safety assessments included heart rate monitoring and changes in 12-lead ECG variables (including heart rate and PR, QRS and QT intervals). These assessments were performed prior to the first dose and then hourly for 6 hours. Day 1 heart rate discharge criteria were the following: ECG-derived resting heart rate more than 45 beats per minute (bpm), and if heart rate was less than 50 bpm it could not be the lowest value post dose; systolic blood pressure (BP) more than 90 mm Hg; QT interval corrected by Fredericia's formula <500 ms; no persistent ECG abnormality (eg, atrioventricular block second degree or higher) or ongoing AE requiring continued hospitalisation. In addition, 24-hour Holter ECG values were assessed. Safety areas of interest, known through clinical experience to be a class effect of S1P receptor modulators, included: cardiovascular effects including heart rate (on day 1), PR interval, systolic and diastolic BP, pulmonary function, immunomodulation including malignancies and infections, macular oedema, liver function test elevation and teratogenicity. The predefined stopping criteria for safety areas of interest are summarised in online supplementary file 3.
Outcome measures
The primary PD endpoint was change in total lymphocyte count from baseline to EOT. Other study endpoints were changes in total lymphocyte count from baseline to each on-treatment assessment and at EOS; treatment-emergent AEs (TEAEs); SAEs; AEs of special interest (AESIs; (defined as per safety area of interest) to include the anticipated risks of treatment with cenerimod, known class effects, or the events related to SLE comorbidities (eg, cardiovascular AEs)); AEs representing a clinical manifestation of an SLE flare (in the investigator’s opinion); and AEs leading to treatment discontinuation (list of AESIs and additional safety endpoints: online supplementary file 4).
Exploratory evaluations of disease activity included changes from baseline to each post-baseline asse ssment in the mSLEDAI-2K score and in the mucocutaneous and/or musculoskeletal SLEDAI-2K subscore. Exploratory biomarker endpoints included changes in anti-dsDNA and blood lymphocyte subsets from baseline to EOT and EOS. Additional exploratory endpoints are described in online supplementary file 5.
Statistical analysis
Based on assumptions from phase I study results,20 a sample size of 64 patients (12 in each cenerimod dose group and 16 in the placebo group) was deemed adequate to provide an average power of at least 90% to show a significant dose–response relationship on lymphocyte count at a one-sided alpha significance level of 5%. This assumed no lymphocyte count reduction from baseline for placebo, and a maximum 70% reduction for any cenerimod dose. For statistical analysis, all four cenerimod treatment groups were compared with combined data from the placebo groups in parts A and B.
To assess any change in total lymphocyte count, an optimised contrast test according to the Multiple Comparison Procedure and Modelling (MCP-Mod) approach22 for each considered dose–response model was used. Five models were prespecified for consideration in MCP-Mod analyses: maximum effect (Emax) curves with 50% of the effective dose (ED50) at 0.2, 0.4 and 1 mg, quadratic curve with Emax at 3 mg and sigmoid-Emax curve with ED50 at 0.4 mg and ED95 at 2 mg. Multiplicity-adjusted p-values were calculated using the Dunnett’s test. PD effects were analysed based on pairwise comparisons of reduction in total lymphocyte count from baseline for each cenerimod dose level with placebo, using an analysis of covariance (ANCOVA) model, adjusted for baseline total lymphocyte count. Statistical testing was based on a two-sided significance level of 5%.
Exploratory analyses of the change from baseline in mSLEDAI-2K score and SLEDAI-2K mucocutaneous and/or musculoskeletal score were performed using an ANCOVA, with treatment group and baseline score as factors. Mean treatment differences for each cenerimod dose compared with placebo and their corresponding two-sided 95% CIs were provided.
In general, analyses were conducted on the full analysis set (FAS) of all 67 randomised patients. Primary PD analyses were initially performed using the PD set, which included all patients who had received study treatment for at least 21 days and had valid baseline and post-baseline total lymphocyte count data. Consequently, the PD set excluded three patients from the FAS: two from the cenerimod 1 mg group and one from the placebo group. Furthermore, Ctrough levels were discovered to be low, or below the lower limit of quantification (BLQ), in four patients randomised to the cenerimod 4 mg group, a finding incompatible with compliance with study treatment. These patients were excluded from the PD set to form a post hoc modified PD (mPD) set. Exploratory analyses on disease activity and biomarkers used the mPD set in addition to the FAS. All 67 randomised patients (ie, the FAS) reported receiving at least one dose of study treatment and were included in the safety set.
Results
Study population
Between 1 June 2015 and 28 February 2017, 105 patients were screened and 67 were randomised to receive study treatment: 49 in part A (randomised 1:1:1:1 to receive cenerimod 0.5, 1, 2 mg or placebo) and 18 in part B (randomised 3:1 to receive cenerimod 4 mg or placebo; figure 1).
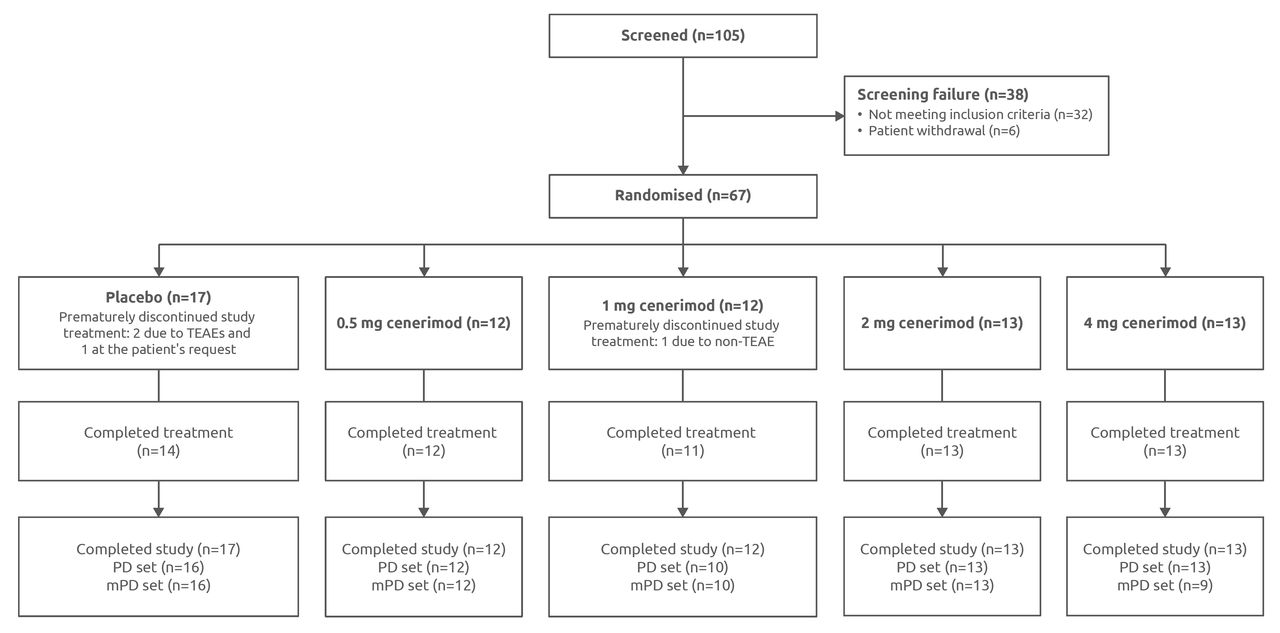
Study profile. FAS and safety set (N=67); patients excluded from the PD set (<21 days of study treatment or missing a baseline or a post-baseline lymphocyte count: n=3); patients excluded from the mPD set (cenerimod plasma concentrations that were undetectable at week 4 or later time points: n=4). Patients were considered to have completed the study if they attended the EOS study visit 6 weeks after study treatment discontinuation. EOS, end of study; FAS, full analysis set; mPD, modified pharmacodynamics; PD, pharmacodynamics; TEAE, treatment-emergent adverse event.
Demographics and baseline characteristics of the study population are shown in table 1. Overall, 61 (91%) patients were female, 65 (97%) were Caucasian and 2 (3%) were African-American. Both African-American patients were in the placebo group. The median times from first SLE symptom and SLE diagnosis varied between groups, with the lowest median values in the cenerimod 0.5 mg group (3.7 and 2.4 years, respectively) and the highest median values in the cenerimod 1 mg group (8.2 and 6.2 years, respectively). Background medication was predominantly oral corticosteroids, antimalarials and/or immunosuppressants.
Demographics and baseline characteristics of study participants
Change in lymphocyte count
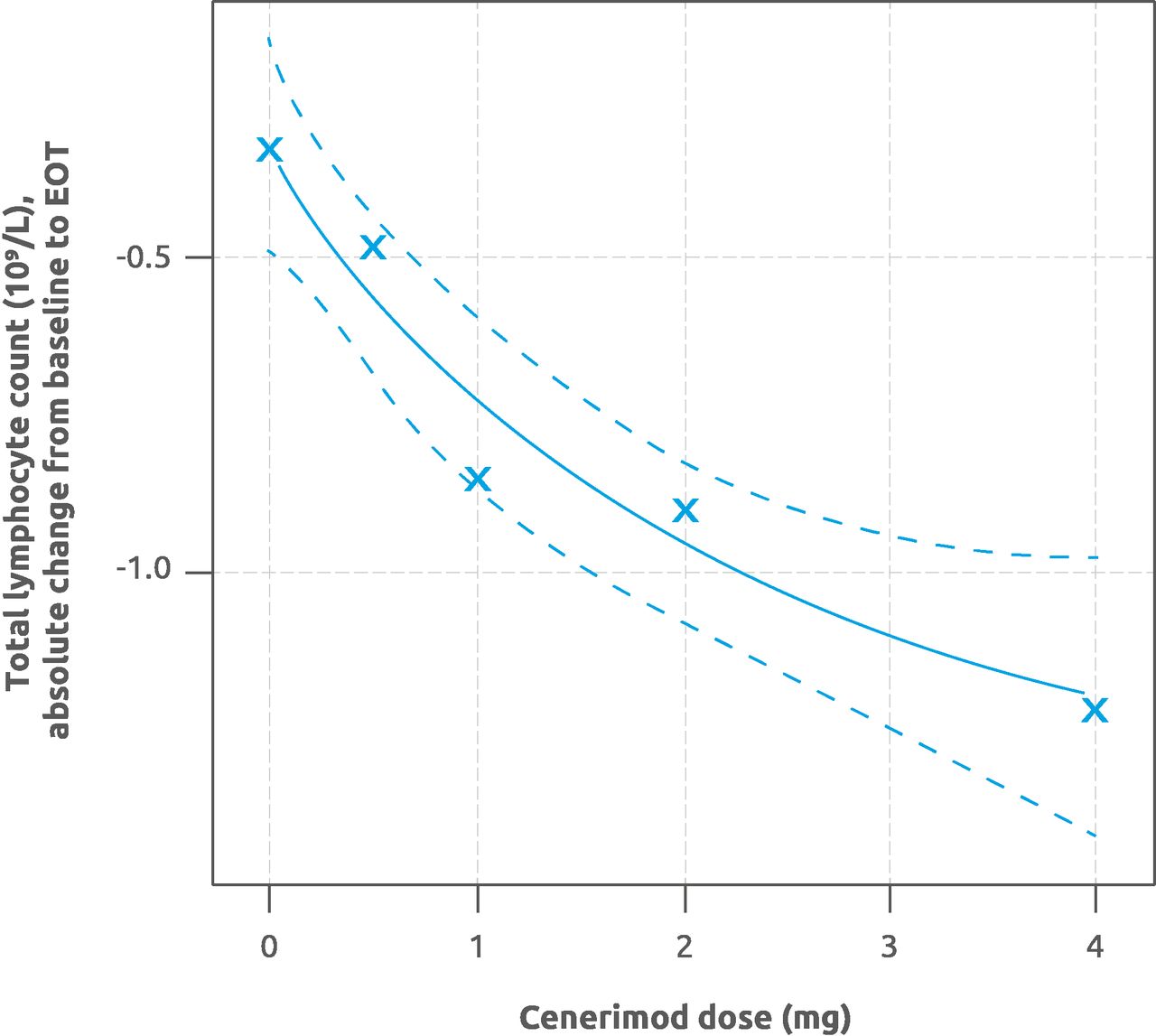
A statistically significant dose–response relationship for change from baseline to EOT in total lymphocyte count was established in the mPD and PD sets, such that all five dose–response models were statistically significant (mPD set p<0.001 and PD set p<0.005, for all five models; figure 2).

Estimation of the dose–response relationship for absolute change in total lymphocyte count from baseline to EOT. The MCP-Mod analysis was performed for each of the five considered dose–response models. Solid line shows the maximum effect (Emax) dose–response curve, and dotted lines show the 95% CI, related to the model with the highest t-statistic. Crosses indicate the measured (observed) absolute change from baseline to EOT. Modified PD set (n=60). EOT, end of treatment; MCP-Mod, Multiple Comparison Procedure and Modelling; PD, pharmacodynamics.
Analysis of the mPD set showed a statistically significant greater mean reduction (±SD) in total lymphocyte count from baseline to EOT with cenerimod 1 mg (0.96±0.68×109/L), 2 mg (0.86±0.61×109/L) and 4 mg (1.48±0.73×109/L) compared with placebo (0.32±0.72×109/L). The mean reduction from baseline to EOT with cenerimod 0.5 mg (0.26±0.48×109/L) was not statistically significant. Average percentage changes in total lymphocyte count from baseline to EOT for the mPD set were −12% (0.5 mg), −48% (1 mg), −52% (2 mg), −69% (4 mg) and −5% (placebo). Analysis of the PD set showed a lower mean reduction (±SD) in total lymphocyte count from baseline to EOT in the 4 mg (0.87±1.24×109/L) group, consistent with low or BLQ Ctrough in four subjects.
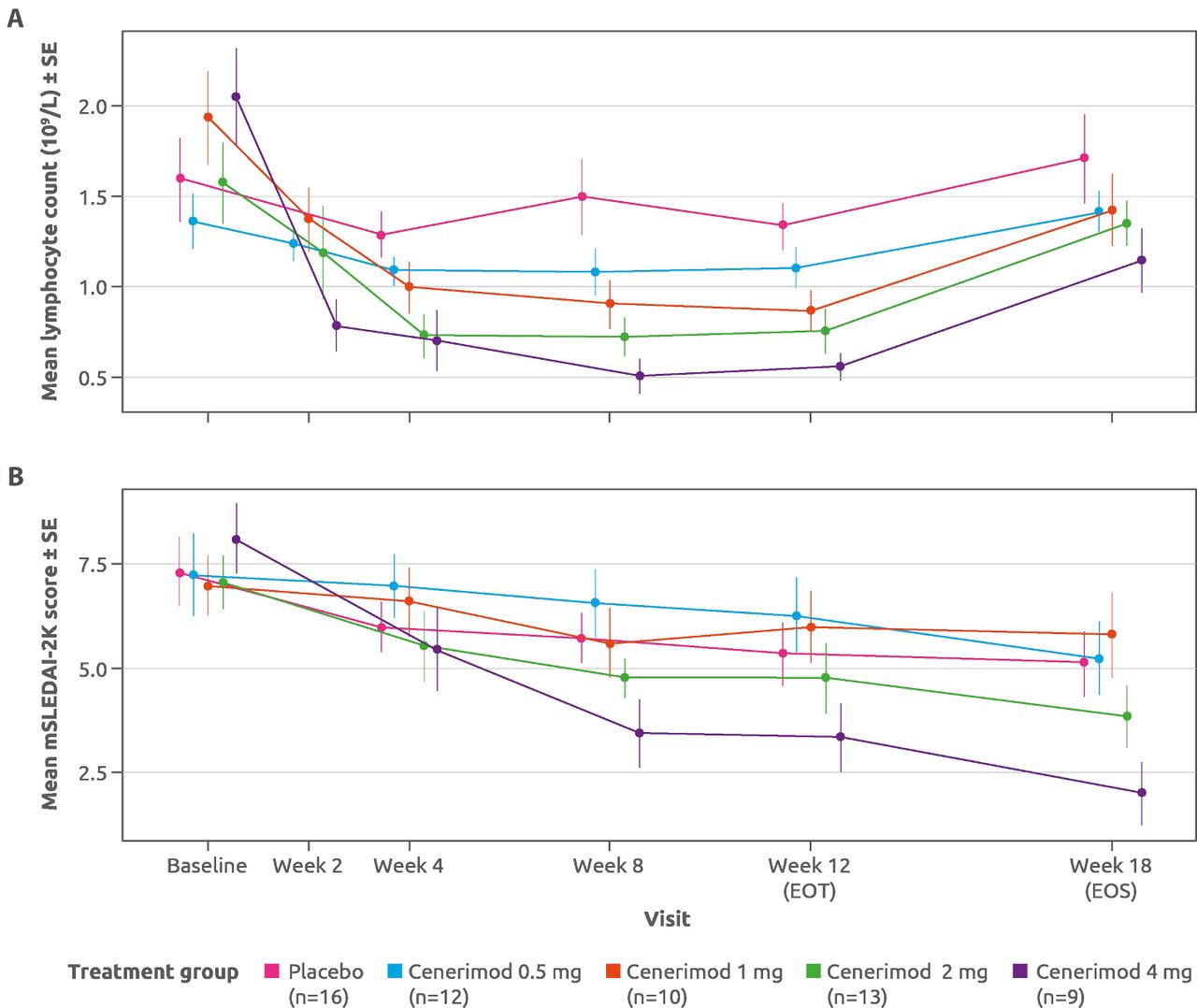
Decreases in mean lymphocyte counts were observed in all cenerimod groups compared with placebo at all on-treatment assessments. This decrease was evident at week 2 and plateaued by week 8, before returning toward baseline values at EOS (figure 3).
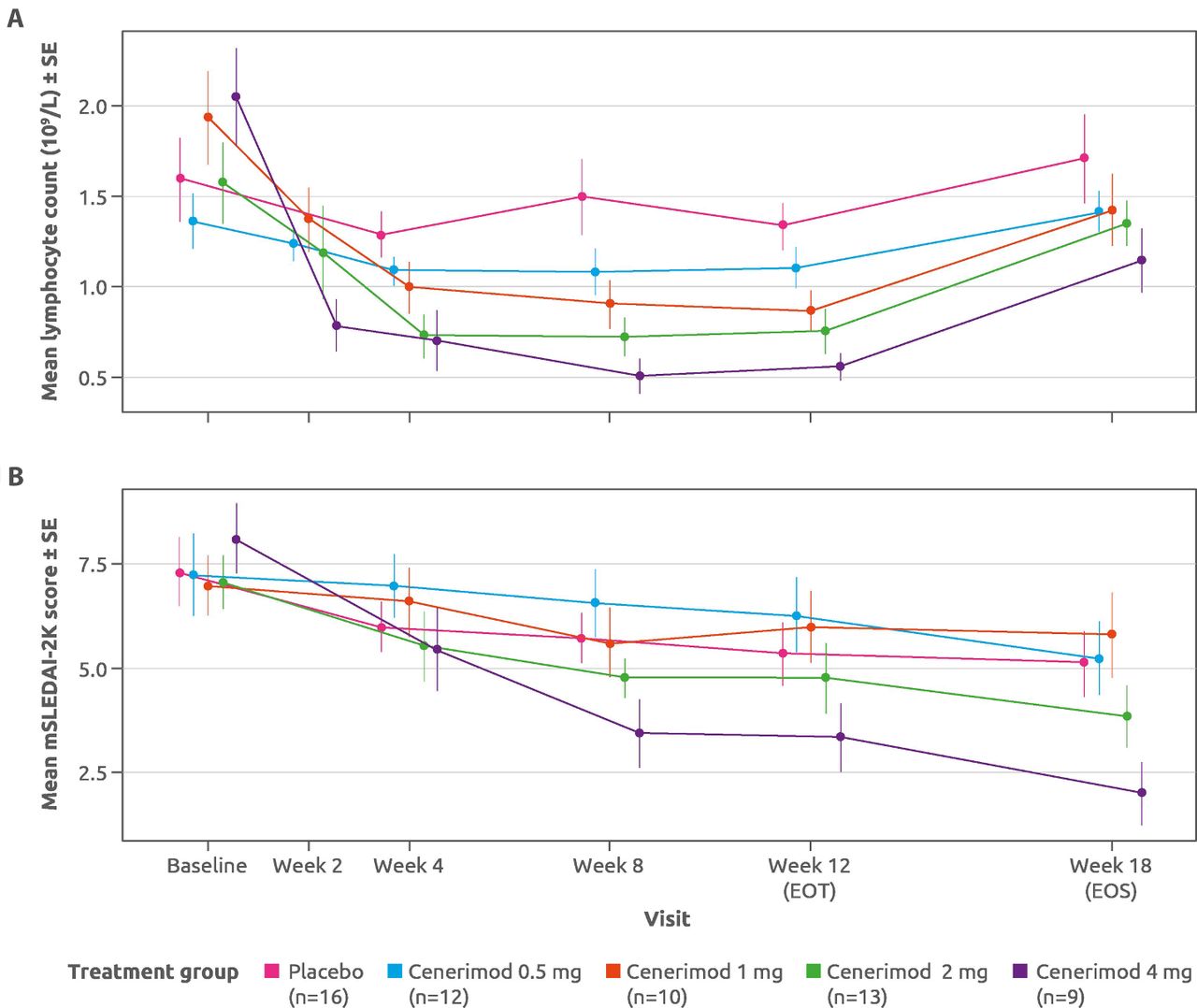
Change in (A) mean lymphocyte count and (B) mean mSLEDAI-2K scores for placebo-treated and cenerimod-treated patients at all study timepoints from baseline to EOS. mPD set (n=60). EOT, end of treatment; EOS, end of study; mPD, modified pharmacodynamics; mSLEDAI-2K, SLE Disease Activity Index-2000 (modified to exclude leucopenia); SE, standard error.
Changes in disease activity
In an exploratory analysis of efficacy using the mPD set, greater mean (±SD) decreases from baseline to EOT in mSLEDAI-2K scores were seen in both cenerimod 2 mg (2.31±2.93) and 4 mg (4.78±3.23) groups, compared with placebo (1.94±2.54) (figure 3; online supplementary file 6). The estimated placebo-adjusted treatment effect of the 4 mg dose, −2.420 (p=0.0306) at EOT was sustained for at least 6 weeks (ie, EOS −3.234; p=0.0060).
Biomarker results
In the mPD set, the observed changes from baseline to EOT in anti-dsDNA antibodies were −18.23 U/mL in the cenerimod 2 mg group, and −53.78 U/mL in the 4 mg group, compared with +12.88 U/mL in the placebo group (online supplementary file 7). These resulted in estimated treatment effects of −24.77 U/mL in the cenerimod 2mg group (95% CI −67.18 to 17.64; p=0.2468) and of −64.55 U/mL in the cenerimod 4 mg group (95% CI −111.7 to –17.43; p=0.0082).
Consistent dose-related decreases in T and B lymphocyte subsets in blood were observed with cenerimod 1, 2 and 4 mg in the FAS. The mean reduction from baseline in T lymphocyte count at EOT was greater with cenerimod 1 mg (0.57±0.80×109/L), 2 mg (0.86±0.53×109/L) and 4 mg (0.70±0.86×109/L) than with placebo (0.19±0.66×109/L), while mean reduction in the 0.5 mg group (0.12±0.30×109/L) was comparable with placebo (see online supplementary file 8A). The mean reduction from baseline in total B lymphocyte count at EOT was greater with cenerimod 1 mg (0.12±0.21×109/L), 2 mg (0.12±0.09×109/L) and 4 mg (0.11±0.16×109/L) than with placebo (0.03±0.07×109/L), while mean reduction in the 0.5 mg group (0.03±0.06×109/L) was similar to placebo (see online supplementary file 8B).
PK results
For all cenerimod groups, Ctrough increased until steady-state conditions were reached at approximately week 4, although the large variability observed in the cenerimod 4 mg group led to fluctuations in Ctrough between week 4 and week 12. At the EOS visit, Ctrough were BLQ for all patients in the cenerimod 0.5, 1 and 2 mg groups and for two patients in the cenerimod 4 mg group (figure 4).
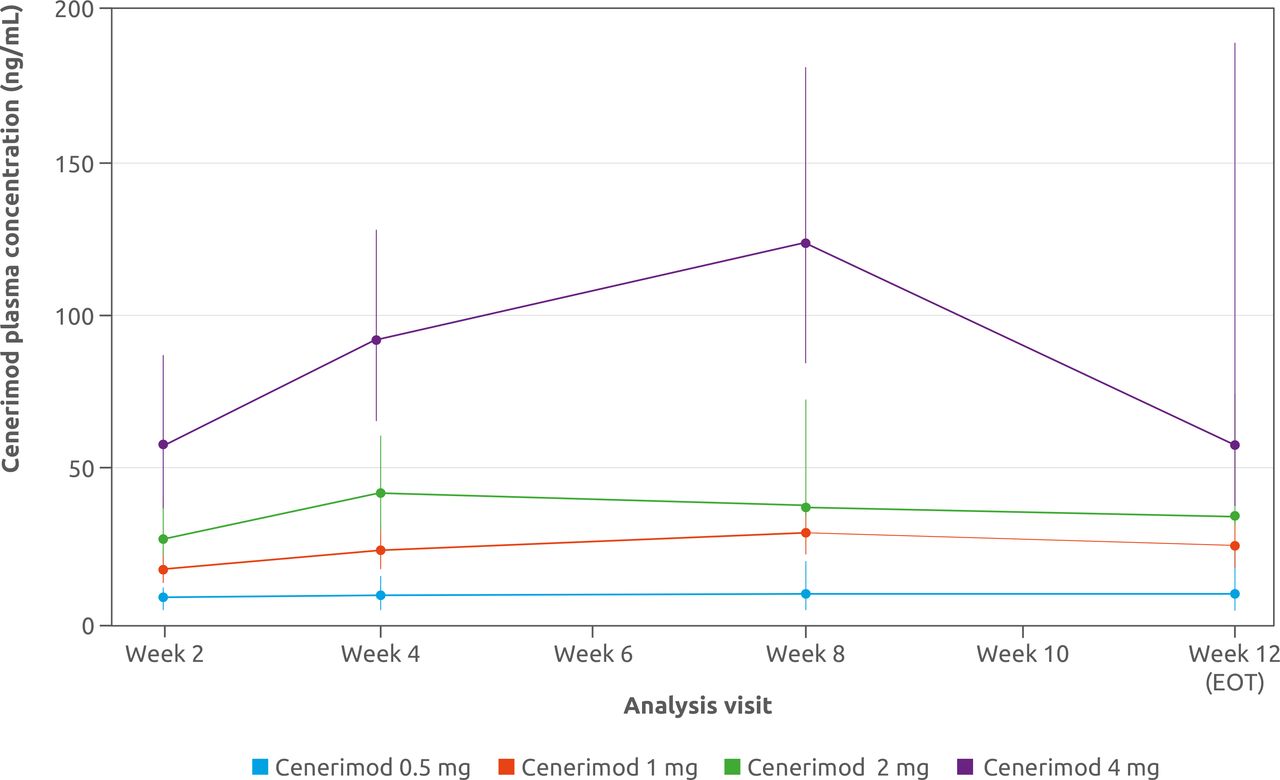
Change in cenerimod plasma concentration for cenerimod-treated patients from baseline to EOT (week 12). Data are presented as geometric mean with 95% CI. mPD set (n=60). EOT, end of treatment; mPD, modified pharmacodynamics.
Safety and tolerability
Cenerimod treatment was well tolerated at all doses tested (analysed for the safety set). The incidence of TEAEs was similar among cenerimod doses and was numerically lower than in the placebo group (cenerimod 0.5 mg: 41.7%; 1 mg: 41.7%; 2 mg: 46.2%; 4 mg: 38.5% and placebo: 58.8%; table 2). Drug-related AEs, considered by the investigator to be related to study treatment, occurred in similar numbers of patients across the placebo and cenerimod treatment groups, and there was no evidence of dose dependency. No drug-related SAEs were reported (table 2).
Treatment-emergent adverse events
In total, three patients discontinued because of AEs. One patient receiving placebo developed severe treatment-emergent SAEs (cholecystitis chronic, pancreatitis chronic (twice) and postcholecystectomy syndrome), which led to discontinuation on day 34. This patient’s SAEs were judged to be a clinical manifestation of an SLE flare unrelated to study treatment. Another patient receiving placebo discontinued the study after a TEAE of dyspepsia on day 22. One patient, receiving cenerimod 1 mg, was diagnosed with a non-TEAE of severe autoimmune hepatitis pre dose on day 1. This AE was judged by the investigator to be related to an SLE flare and led to discontinuation on day 9.
Less than 10% of patients experienced an AESI (n=6; 9%); five patients had liver-related AEs and one had a pulmonary-related AE. Numbers of reported AESIs were comparable across the cenerimod and placebo treatment groups with no evidence of dose dependency (table 2 and online supplementary file 9). All laboratory values were below the defined thresholds for marked abnormality. The pulmonary-related AE was non-serious severe pneumonitis, which occurred in a patient receiving cenerimod 1 mg and was judged by the investigator to be unrelated to study treatment.
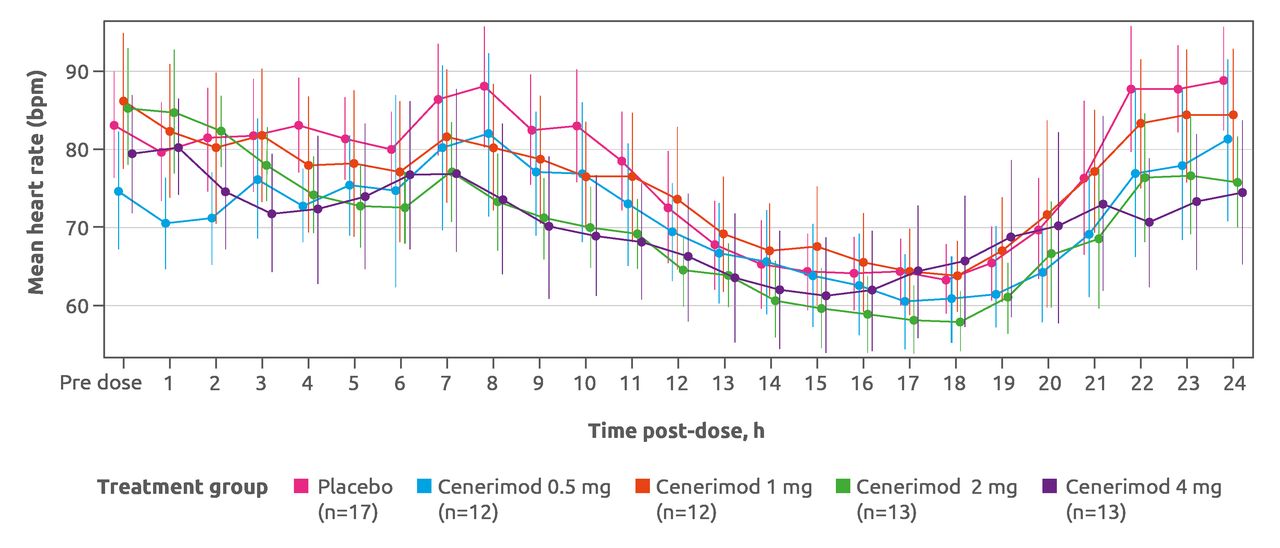
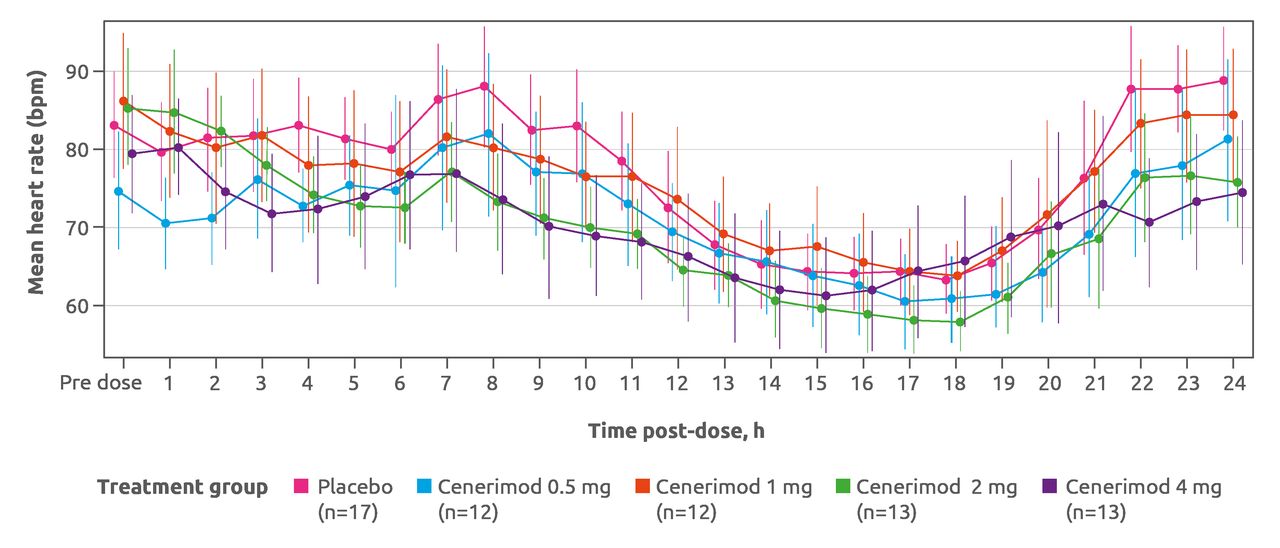
After the first dosing, cenerimod induced a dose-dependent, transient and minimal decrease in heart rate (figure 5). On day 1, 12-lead ECG measurements hourly from pre dose to 6 hour post dose revealed that no patient had a heart rate lower than 40 bpm at any time after baseline, and all patients had systolic BP higher than 90 mm Hg. No patient failed to meet the heart rate or BP discharge criteria at 6 hours. Cenerimod did not affect PR or QRS intervals, although one patient (receiving placebo) had an abnormal PR interval of more than 200 ms. From the week 2 visit onward, no evidence for an effect with cenerimod was seen in any of the 12-lead ECG variables. Mean and median changes from baseline in supine systolic and diastolic BP over the first 6 hours on day 1 showed no difference between cenerimod-treated and placebo-treated patients. No trends could be discerned, and there was no evidence of a dose effect.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hourly heart rate on day 1 monitored by 24-hour Holter ECG. Data shown are mean and 95% CI. Safety set (N=67). bpm, beats per minute
Mean and median changes from baseline in spirometry variables indicated a small decrease in pulmonary function by EOT for cenerimod-treated patients that was not clearly dose related (see online supplementary file 10). The largest median decrease from baseline to EOT in absolute FEV1 and FVC was observed in the cenerimod 2 mg group (−0.17 and −0.15 L, respectively) compared with placebo (0.00 and −0.06 L, respectively). Decreases of more than 15% from baseline to EOT in FEV1 were observed in five patients (two patients in the placebo group, one in each of the cenerimod 0.5, 1 and 2 mg groups). A decrease above 15% decrease from baseline to EOT in FVC was also observed in five patients (two in the placebo group, two in the cenerimod 0.5 mg and one in the 4 mg group). No decreases in FEV1 or FVC of more than 15% from baseline to EOT were observed at more than one timepoint in any of the cenerimod-treated patients. There was no evidence of a dose-related effect and none of these decreases or changes from baseline to EOT were associated with clinical symptoms.
Cenerimod treatment did not affect rates of infection, physical findings or body weight, nor did it lead to clinically significant ophthalmological disorders. There were no trends in changes in supine systolic and diastolic BP between cenerimod-treated and placebo-treated patients at any assessment during the treatment duration. None of the safety events met the protocol predefined safety stopping criteria.
Discussion
The current study is the first to investigate the PD, PK and safety of the oral selective S1P1 receptor modulator, cenerimod, in patients with SLE. In this 12-week, randomised, double-blind, placebo-controlled trial, daily oral doses of cenerimod dose-dependently reduced circulating lymphocytes, with reductions evident as early as week 2. These findings in patients with SLE confirm non-clinical findings19 and the mode of action of cenerimod as a functional antagonist of S1P, preventing lymphocyte egress from lymphoid tissues into the circulation. In addition, since cenerimod is characterised by a long half-life, and therefore accumulates,20 Ctrough is expected to accurately reflect drug exposure, indicating that steady-state conditions are reached after approximately 4 weeks in patients with SLE, in keeping with previous findings in healthy subjects. Exposure to cenerimod was comparable to that of healthy participants,20 suggesting that the PK/PD profile established in healthy participants also applies to patients with SLE.
Findings from this 12-week study suggest that cenerimod has the potential to reduce disease activity in a dose-dependent manner in the first 3 months of treatment. Numerical reductions from baseline to EOT in mSLEDAI-2K score and mucocutaneous SLEDAI-2K subscore were observed across cenerimod groups, with greater decreases in mSLEDAI-2K scores in the cenerimod 2 and 4 mg groups. In addition, a pronounced decrease in the SLE biomarker, anti-dsDNA, was seen with the two higher cenerimod doses when compared with placebo.
Cenerimod was well tolerated at all doses tested, showing no evidence of dose-dependent toxicity. The incidence of AEs in cenerimod-treated patients was numerically lower than placebo, and the one patient who had a treatment-emergent SAE was in the placebo group. Dose initiation was safe as shown by the ECG data and by the fact that no patients failed to meet the criteria for discharge on day 1. Furthermore, decreases in FEV1 and FVC at EOT were small and clinically non-significant, with no evidence of a dose effect.
S1P modulators are known to exhibit concentration-dependent reductions in heart rate.18 23 24 This effect reduces with continued exposure (ie, tolerance develops), which is attributed to receptor internalisation and desensitisation of the S1P receptor system following repeated dosing. Development of tolerance allows for gradual uptitration to the desired dose, and the titration scheme can be optimised to reduce the effect on heart rate.25 Cenerimod exhibits a moderate first-dose decrease in heart rate. Its slow accumulation offers a gradual desensitisation per se without the need for uptitration. The desired dose can be given from the first day of treatment, avoiding complications arising from uptitration schedules (eg, usage of blisters with prescribed intake sequence or having to restart uptitration if doses are missed). Nevertheless, underlying cardiac abnormalities should be taken into consideration before initiating cenerimod treatment.
A limitation of this study includes the short treatment duration, which made assessment of the durability of therapy benefits impossible. This will be important for future clinical trials because SLE is a chronic disorder with a waxing and waning disease course. In addition, patients with more severe disease (SLEDAI-2K score >12) at baseline were not eligible for the study. Despite the short treatment duration and patient population having been restricted to those with mild-to-moderate disease, the clinical data presented underscore the potential of cenerimod to specifically modulate SLE disease pathophysiology and to translate into clinical efficacy.26
In conclusion, cenerimod has the potential to be a promising new therapeutic approach for patients with SLE and has an acceptable safety profile with minimal, non-clinically relevant cardiovascular effects. These findings warrant further investigation of cenerimod in a larger patient population and with a longer treatment duration to determine the extent of its efficacy as a treatment for SLE. A 500-patient phase IIb, randomised, dose-finding study was initiated in December 2018 to evaluate the efficacy and safety of cenerimod, in addition to background therapy, in patients with moderate-to-severe SLE (ClinicalTrials.gov: NCT03742037).
Acknowledgments
The authors thank the investigators, staff and the patients of the study for their valuable contributions, as well as Marilia Pozzobon da Silva, MD, of Idorsia Pharmaceuticals, Allschwil, Switzerland, for her help in revising the manuscript. The authors also thank Zoe Kelly, PhD, of InterComm International, Cambridge, UK, for medical writing and editorial support, which was funded by Idorsia Pharmaceuticals.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Lay summary
Disclaimer : This is a summary of a scientific article written by a medical professional (“the Original Article”). The Summary is written to assist non medically trained readers to understand general points of the Original Article. It is supplied “as is” without any warranty. You should note that the Original Article (and Summary) may not be fully relevant nor accurate as medical science is constantly changing and errors can occur. It is therefore very important that readers not rely on the content in the Summary and consult their medical professionals for all aspects of their health care and only rely on the Summary if directed to do so by their medical professional. Please view our full Website Terms and Conditions.
Copyright: © Author(s) (or their employer(s)) 2019. This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0.
Footnotes
Contributors VH reviewed and interpreted the data. AB and SS were involved in patient recruitment, conduct of the study and acquisition of data. P-EJ analysed and provided interpretation of the PK data. PC provided oversight, review and interpretation of the statistical analyses, and carried out further analyses. All authors were involved in drafting the article and revising it critically for important intellectual content, and all authors approved the final version to be submitted.
Funding This study was sponsored by Actelion Pharmaceuticals. Study sponsorship was transferred to Idorsia Pharmaceuticals in July 2018. These sponsors participated in the design and conduct of the study, collection, management, analysis and interpretation of the data.
Competing interests VH, P-EJ and PC are employees and shareholders of Idorsia.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.