Article Text

Abstract
Objective Changes in the care of patients with SLE dictate a re-evaluation of its natural history and risk factors for disease deterioration and damage accrual. We sought to decipher factors predictive of a deterioration in phenotype (‘transition’) in patients initially presenting with non-severe disease.
Methods Patients from the ‘Attikon’ cohort with disease duration ≥1 year were included. Disease at diagnosis was categorised as mild, moderate or severe, based on the British Isles Lupus Assessment Group manifestations and physician judgement. ‘Transition’ in severity was defined as an increase in category of severity at any time from diagnosis to last follow-up. Multivariable logistic regression was performed to identify baseline factors associated with this transition.
Results 462 patients were followed for a median (IQR) of 36 (120) months. At diagnosis, more than half (56.5%) had a mild phenotype. During disease course, transition to more severe forms was seen in 44.2%, resulting in comparable distribution among severity patterns at last follow-up (mild 28.4%, moderate 33.1%, severe 38.5%). Neuropsychiatric involvement at onset (OR 6.33, 95% CI 1.22 to 32.67), male sex (OR 4.53, 95% CI 1.23 to 16.60) and longer disease duration (OR 1.09 per 1 year, 95% CI 1.04 to 1.14) were independently associated with transition from mild or moderate to severe disease. Patients with disease duration ≥3 years who progressed to more severe disease had more than 20-fold increased risk to accrue irreversible damage.
Conclusion Almost half of patients with initially non-severe disease progress to more severe forms of SLE, especially men and patients with positive anti-double-stranded DNA or neuropsychiatric involvement at onset. These data may have implications for the management of milder forms of lupus.
- systemic lupus erythematosus
- epidemiology
- antiphospholipid antibodies
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE is a systemic autoimmune disease with protean clinical manifestations and an unpredictable course.1 Although prognosis has significantly improved over the years due to earlier diagnosis and more effective treatments, patients with SLE still demonstrate increased mortality and morbidity compared with the general population.2 Patients’ phenotype at disease onset may vary from mild to severe or life-threatening,3 4 with striking differences among patients from different racial backgrounds. Lupus nephritis (LN) is more common in Hispanics and African–Americans,5 6 the latter also exhibiting an up to twofold increased risk of neuropsychiatric involvement, compared with Caucasians.7 8
Several cohort studies around the world have documented the natural history and morbidity of the disease, contributing substantially to increased awareness.9 More recently, emphasis has been put on the patterns of disease activity and targets of therapy, with remission and low disease activity emerging as new frontiers.10 Moreover, management recommendations have attempted to decrease the heterogeneity in lupus care, by providing evidence-based and expert opinion-based guidance.11 However, among patients who present with a certain phenotype, there is a paucity of data regarding potential changes of severity over time, that is, whether the disease will remain mild throughout its course or progress to a more severe form. Such data may have clinical and therapeutic implications for early disease.
The aim of this study was to describe the severity patterns of a Caucasian SLE cohort in a tertiary SLE referral centre, based at ‘Attikon’ University Hospital, Athens, Greece. We explored possible baseline prognostic factors related to a ‘transition in severity’ as well as cumulative damage accrual over the course of the disease. Our data suggest that, despite significant advances in therapy, transition of disease occurs in a considerable proportion of patients.
Patients and methods
Patients and clinical assessment
‘Attikon’ University Hospital is a tertiary centre located in a large urban area of Western Attica, responsible for the healthcare of close to two million local residents. An SLE cohort was initiated in January 2014 to include all patients diagnosed with SLE who had a regular follow-up as outpatients. The ‘Attikon’ lupus cohort consists of a ‘prevalent cohort’ (patients with an SLE diagnosis prior to the establishment of the patient registry) and an ‘inception’ cohort (patients followed from diagnosis onwards).12 A standardised data set, including demographics and clinical and laboratory features of the disease, is completed for each patient at first visit and every follow-up. All immunosuppressive/immunomodulatory drugs administered for the treatment of SLE are also documented, including current treatment (ie, at most recent visit) and past medications. Patient enrolment for the purpose of the study was completed in January 2019.
Patients with SLE fulfilling the American College of Rheumatology (ACR)13 and/or Systemic Lupus International Collaborating Clinics (SLICC) criteria14 and who had disease duration ≥1 year were included in this study. LN was defined according to SLE classification criteria and/or kidney biopsy.13 14 A diagnosis of primary neuropsychiatric SLE (NPSLE) was established according to the ACR definitions,15 following a combination of expert physician judgement (DTB, AF).16 17 For patients enrolled in the cohort after the neuropsychiatric manifestation had occurred, attribution to SLE or not was based on patient history and all available data (taking into account a variety of risk factors for NPSLE at the time of neuropsychiatric involvement, ie, antiphospholipid antibodies (aPL), prior neuropsychiatric manifestation, generalised disease activity),16–18 or was considered as ‘uncertain’. For the definition of childhood-onset SLE, a cut-off of 17 years was used,19 whereas onset after 50 years was defined as late-onset SLE. For the assessment of damage, the SLICC Damage Index (SDI)20 was captured yearly for each patient.
Definitions of disease severity and ‘transition’
For the purpose of this study, the phenotype of SLE was categorised as mild, moderate or severe across two timepoints: diagnosis and most recent follow-up. For patients enrolled in the cohort after the disease had been diagnosed (prevalent cohort), phenotype at diagnosis was based on patient history and all available data on patient file. Medical charts of all patients were scrutinised to detect incident manifestations (at any timepoint across the disease course) from individual organ systems. Stratification of disease during the course of the disease was determined by expert physician (DTB, AF) based on a structured assessment that took into account (1) the presence of disease manifestations graded in severity according to the British Isles Lupus Assessment Group (BILAG) 2004 index glossary21 and (2) all treatments received by patients. Specifically, severe disease was defined as (1) severe SLE manifestation from at least one organ according to the BILAG glossary and/or (2) treatment with cyclophosphamide or rituximab (for any manifestation, other than arthritis) at any time over disease course.8 Mild disease was defined as (1) mild manifestations according to the BILAG glossary, (2) absence of any major organ involvement and (3) maximum treatment with the following: oral glucocorticoids (GC) ≤10 mg/day (prednisone equivalent) or intramuscular GC and/or hydroxychloroquine (HCQ), at any time during disease course. Patients falling between these two definitions were classified as moderate disease. Patients were assessed at each visit for possible transition to a more severe form of the disease (ie, from mild to moderate/severe, or from moderate to severe). As this ‘transition in severity’ was the primary outcome, patients with severe lupus at diagnosis were excluded from this analysis.
Statistical analysis
Descriptive statistics were undertaken for continuous variables, and mean/SD or median/IQR values were calculated for normally and non-normally distributed variables, respectively. χ2 or Fisher’s exact test was used to compare categorical variables, and Student’s t-test or non-parametric Mann-Whitney U test was used to compare continuous variables, as appropriate.
Logistic regression models were used to identify factors that were independently associated with ‘transition in severity’ and damage accrual. Because patients with initially mild disease may progress to either moderate or severe disease, while those with initially moderate only to severe disease, two different regression analyses were performed, for the identification of baseline risk factors for (1) transition from mild to moderate disease and (2) transition from mild or moderate to severe disease. All variables with a p value <0.20 in univariable analyses qualified for further analysis in age-adjusted multivariable models. P values, ORs and their 95% CI were computed. A stepwise backward selection was performed to eliminate non-significant factors. Model selection and checking were based on tests for linearity, interactions and goodness of fit. For comparisons, statistical significance was indicated as a two-sided p<0.05. All statistical analyses were performed using SPSS V.25.0I.
Information about the study along with the consent form was provided to patients with SLE. All participants signed the informed consent forms.
Results
Patient characteristics
A total of 462 patients, all Caucasians, were included in the study. The mean (SD) age at lupus diagnosis was 37.3 (15.2) years, with a female to male ratio of ~9:1, and the median (IQR) disease duration to last follow-up was 36 (120) months. Fifty (10.8%) patients were diagnosed with childhood-onset SLE and 98 patients (21.2%) with late-onset SLE.
The most common clinical manifestations at diagnosis were inflammatory arthritis (72.7%), acute cutaneous lupus (63.2%, mainly malar rash and photosensitive rash), leucopenia (22.5%) and non-scarring alopecia (22.5%). LN was manifest at onset in 44 (9.5%) patients, while 61 (13.2%) more patients developed renal involvement during follow-up, reaching an overall prevalence of 22.7%. There were 112 primary neuropsychiatric manifestations observed in 86 patients (18.6% of total population). Approximately 60% of patients with NPSLE (51 of 86) had at least one SLE-related neuropsychiatric manifestation at the time of diagnosis, while 35 (39.7%) patients manifested NPSLE during follow-up. Clinical and serological items are summarised in table 1.
Clinical and serological items of SLE at the time of diagnosis and cumulatively
The vast majority of patients in our cohort had received HCQ and oral GC at some point during the course of their disease (95.0% and 98.3%, respectively); at most recent follow-up, the respective percentages were 85.6% and 67.9%. Use of additional immunosuppressive medications is shown in online supplementary figure 1.
Supplemental material
Transition of disease severity over time and predictors
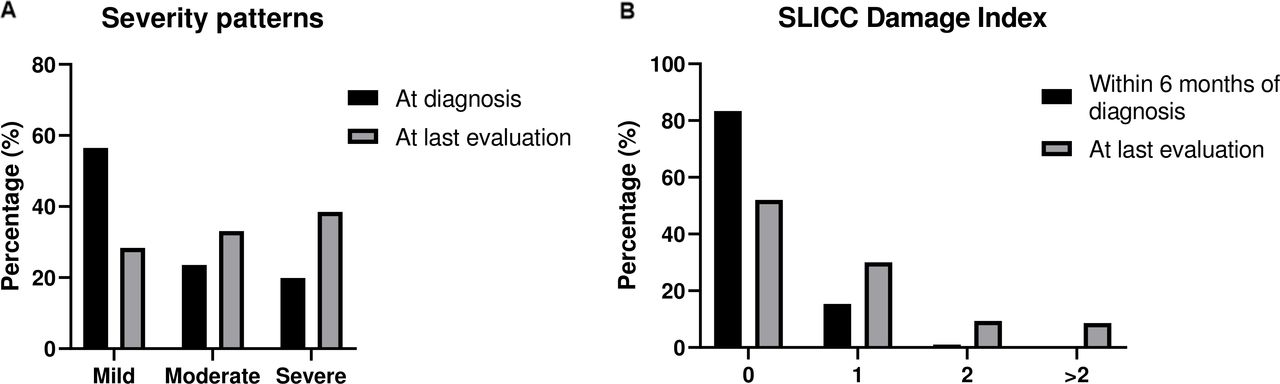
The respective distribution of disease severity at diagnosis and over time is depicted in figure 1A. More than half of patients (261 of 462, 56.5%) initially presented with mild disease. Of them, at last assessment, only 131 (50.2%) patients had retained their mild phenotype, while the remaining had evolved to more severe forms: 76 (29.1%) and 54 (20.7%) developed moderate and severe lupus, respectively. Of patients with initially moderate disease (n=109), 32 (29.4%) progressed to severe SLE, while approximately 20% (n=92) of patients had severe manifestations already at diagnosis. This kinetics resulted in an almost equal distribution among the three severity pattern groups (mild, moderate, severe) at last assessment (figure 1A).
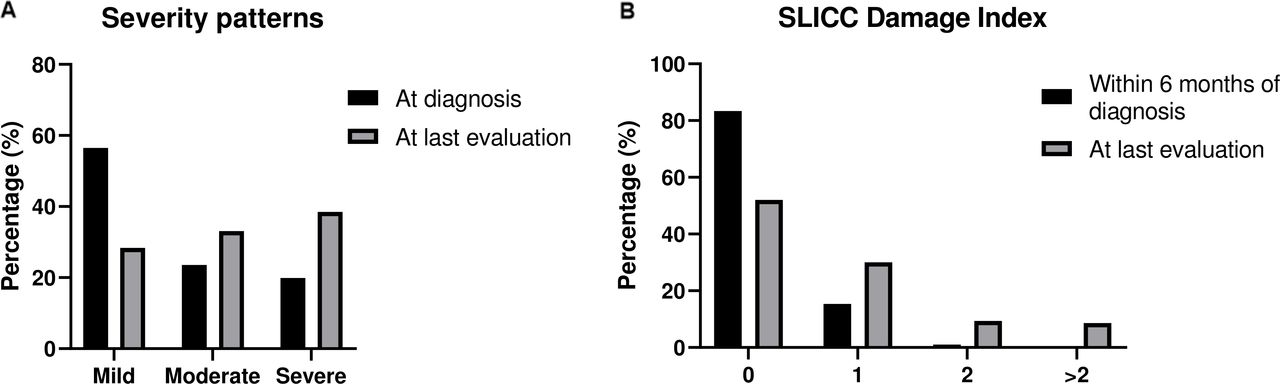
(A) Disease severity patterns of patients with SLE (‘Attikon’ cohort) at disease onset and at last evaluation. (B) Damage accrual of patients with SLE (‘Attikon’ cohort) within 6 months after diagnosis and at last evaluation. SLICC, Systemic Lupus International Collaborating Clinics.
Patients diagnosed initially mild disease (n=261) were analysed to identify baseline factors as predictors of disease transition to a moderate phenotype (table 2). In both univariable and multivariable analyses, positive anti-double-stranded DNA (anti-dsDNA) at diagnosis and disease duration were associated with transition to moderate lupus (OR 2.39, 95% CI 1.07 to 5.32 and 1.05, 95% CI 1.00 to 1.11, respectively). For transition to severe disease, we included patients presenting initially with either mild or moderate disease (n=370). First, the two disease states (mild vs moderate) did not differ in their risk of transition to a severe phenotype (table 3). Factors associated with this transition in multivariable analysis were male sex (OR 4.53, 95% CI 1.23 to 16.60), disease duration (OR 1.09, 95% CI 1.04 to 1.14) and especially neuropsychiatric involvement at onset (OR 6.33, 95% CI 1.22 to 32.67); presence of anti-dsDNA marginally did not reach statistical significance (OR 1.89, 95% CI 0.96 to 3.73). For both transitions (ie, from mild to moderate, as well as from mild/moderate to severe), patients with late-onset SLE showed a trend to retain their initial phenotype compared with patients diagnosed before the age of 50, only in univariable analyses (tables 2 and 3). We also examined whether different baseline characteristics could predict transition to moderate versus severe disease in patients diagnosed initially with mild SLE, but the results did not differ significantly (data not shown).
Baseline features as predictors of phenotype transition from mild to moderate disease
Baseline features as predictors of phenotype transition from mild or moderate to severe disease
To overcome the potential bias of a shorter disease duration in patients who were less likely to progress to more severe forms (either from mild to moderate, or from mild/moderate to severe), we performed a subgroup analysis in patients with a median disease duration shorter than 3 years; the final age-adjusted and sex-adjusted models remained almost identical in terms of statistical significance (data not shown).
Transition in severity in childhood-onset and late-onset SLE
The childhood-onset SLE population exhibited LN approximately twice more commonly (42% vs 20.6%, p=0.001). Transition to more severe disease at last follow-up was detected in 54.1% of patients with childhood-onset SLE compared with 43.6% in adult-onset patients, a difference not reaching statistical significance. No difference between groups was observed in terms of patterns of severity, SDI and major organ involvement (p>0.05). A higher incidence of moderate/severe disease at diagnosis (combined 56.2% vs 40.3%, p=0.005) and a respective lower incidence of transition to more severe forms (19.7% vs 50.7%, p<0.001) were seen in patients with late-onset disease, as compared with ‘non-late-onset’ patients. The latter difference remained significant even after adjusting for disease duration.
Baseline predictors for damage accrual during follow-up
Seventy-six (16.5%) patients had already established damage within 6 months of disease diagnosis, mainly due to neuropsychiatric and thrombotic components of the SDI (online supplementary table 1). After a median (IQR) disease duration of 3 (10) years, 241 (52.2%) patients had still not accrued damage (SDI=0). A high damage index (SDI ≥3) was found in 40 subjects (8.6%) (figure 1B).
Supplemental material
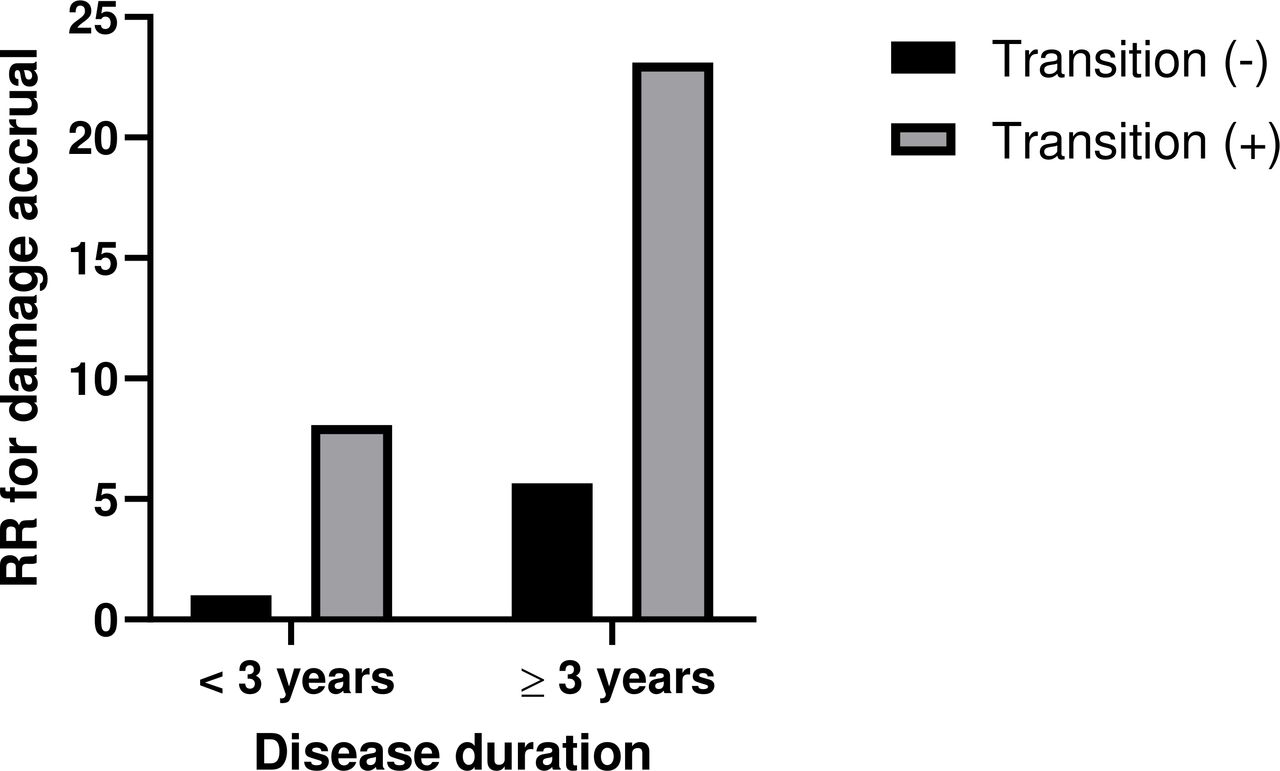
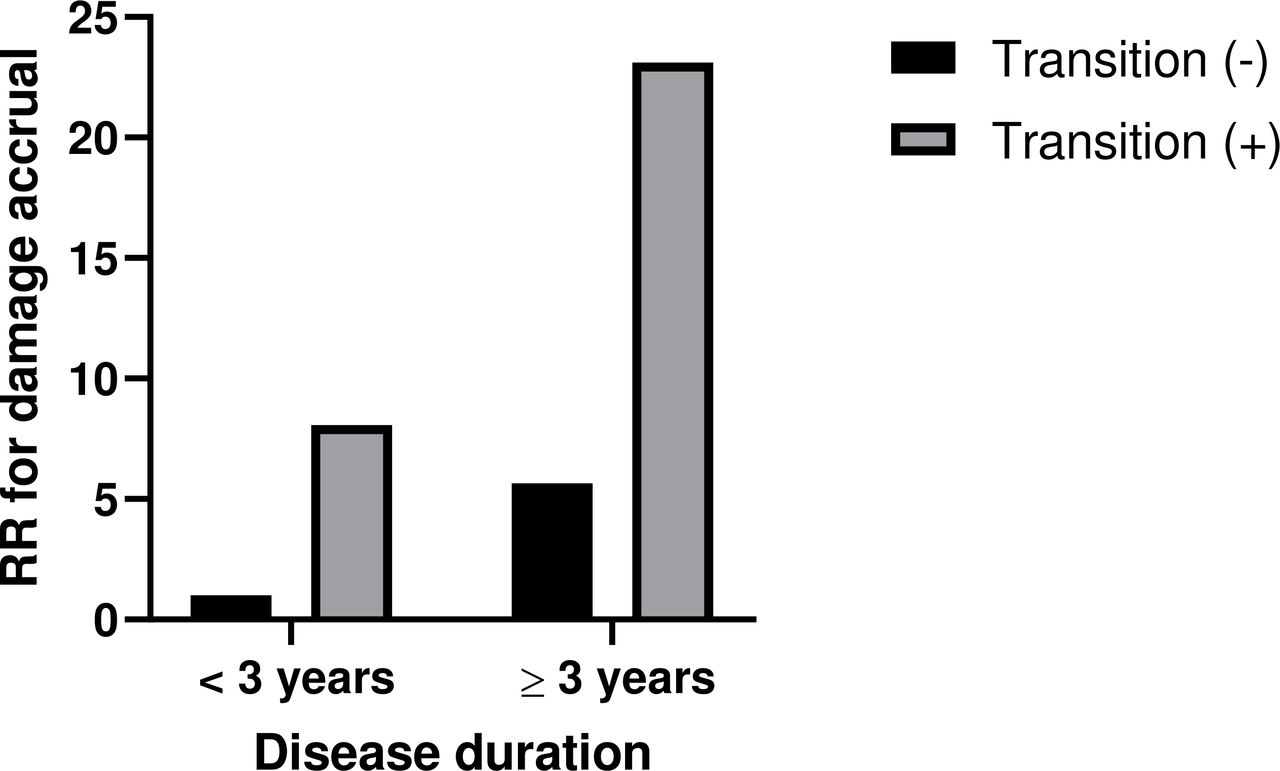
To identify predictors of damage accrual over time in all patients, we performed univariable and multivariable analyses (n=462) (table 4). Univariable analysis revealed comorbidities including hypertension, dyslipidaemia and obesity as predictors of SDI development. Age at diagnosis, disease duration and severity transition were found to be independent predictors of increased SDI in multivariable analysis. As expected, patients who evolved to more severe forms of lupus and patients with longer disease duration exhibited higher risk of damage development (OR 5.66, 95% CI 2.74 to 11.67 and 1.15, 95% CI 1.09 to 1.22, respectively). When disease duration was examined as a binary variable, subjects with longer disease duration (≥3 years) and transition to more severe forms had a 23-fold risk of damage accrual compared with those with preserved disease state and shorter disease duration (figure 2). The presence of positive aPL also conferred a significant risk of damage accrual in our cohort (OR 2.22, 95% CI 1.09 to 4.53).
{kind=link}
{kind=link}
Relative risk (RR) of damage accrual in subjects with different combinations of disease duration and transition compared with those with short disease duration (<3 years) who never progressed to more severe forms of the disease.
Baseline features as predictors for damage accrual
Discussion
The ‘Attikon’ lupus cohort was established in 2014 with the purpose to study the natural history of SLE in a Caucasian population of the modern era. SLE may often follow an unpredictable course; thus, it would be helpful to predict which patients will ultimately develop severe disease necessitating more aggressive treatment. In this study, we aimed to explore factors which could help identify patients who will eventually develop a severe phenotype, although initially presenting with mild or moderate disease. Importantly, to stratify patients in terms of disease severity, we used a combination of BILAG classification and expert judgement, the former being a validated instrument for SLE activity and severity.22 We found that, although approximately 60% of patients present with mild disease at onset, almost 50% of them later progress to a moderate and severe phenotype. These data may have important implications for the management of patients with milder forms of the disease, a subset of which may require closer monitoring.
Following the advent of potent immunosuppressive therapies, the phenotype of rheumatic diseases has changed in certain circumstances, with the prevalence of certain severe manifestations having decreased.23 Most recent cohorts of patients with SLE report rates of LN substantially lower than the ~60% of traditional cohorts, potentially reflecting better disease monitoring and management at the early stages.6 8 24 In this regard, it was important to find that transition to a more severe phenotype is still a reality for a significant proportion of patients. Few studies have examined the temporal characteristics of different lupus manifestations over time. A study undertaken to inform the recently published ACR/European League Against Rheumatism criteria for SLE described disease manifestations at disease onset, but did not report on subsequent follow-ups.4 Also, recent updates from the established Hopkins and Toronto lupus cohorts confirmed that the majority of patients with lupus still tend to follow a relapsing-remitting course25 26; however, whether flares of disease lead to a more severe disease in terms of new organ manifestations was not specified, although both number and severity of flares are known to contribute to damage in lupus.27 28
In a study relevant to our own, Kwon et al29 examined baseline predictors for subsequent development of LN, in patients not presenting initially with renal involvement. Interestingly, anti-dsDNA positivity and younger age at disease onset were independently associated with future LN occurrence, similar to their association with transition to a severe phenotype in our study. These findings strengthen the notion that young, male, anti-dsDNA-positive patients should be under close surveillance for subsequent development of severe disease manifestations. We also found neuropsychiatric involvement at onset to have the strongest association with subsequent transition to severe lupus. Indeed, past neuropsychiatric manifestations have been shown to associate with subsequent occurrence of similar or different neuropsychiatric events and constitute a risk factor for NPSLE.30 31 This is particularly important, as in our cohort we have found increased prevalence of neuropsychiatric involvement (11.5% of patients at disease onset).
Irreversible damage accrual, measured by the SDI, is a milestone in the natural history of SLE, since it has been directly linked to increased mortality.32 33 Importantly, at last follow-up, more than 50% (52.4%) of patients in our cohort still had an SDI of 0. Nevertheless, the median disease duration in patients included in the current study was relatively short (3 years), and a significant proportion (16.5%) already had evidence of damage at diagnosis. Not unexpectedly, we found that transition to a more severe phenotype was independently associated with increased risk for damage, especially with increasing disease duration. In a recent work examining damage trajectories in childhood-onset SLE, major organ involvement was also characterised by a more rapid damage accrual.34 These observations have obvious implications for patients diagnosed at a young age and call for vigilant monitoring and optimal disease control at early disease stages. We also found, in accordance to previous studies, that aPL also contributes independently to damage accrual in SLE.35 36
Our study has several limitations. The ‘Attikon’ lupus cohort consists exclusively of Caucasians; thus, our findings have to be replicated in patient cohorts of different race and ethnicity. Also, in a significant proportion of patients in the prevalent cohort, data regarding history, manifestations and treatments prior to inclusion in the SLE cohort were performed retrospectively. Especially regarding treatments, the specific timing of treatment with each immunosuppressive drug in relation to disease ‘transition’ was not available in all our patients. One could assume that the higher risk of transition in patients with mild disease may be attributable to undertreatment, rather than the natural history of the disease per se. However, more than 95% of patients in our prevalent cohort have been treated with HCQ and GC, which indicates that patients with mild disease had been prescribed appropriate therapy. Notwithstanding the limitation that we lack data regarding adherence to treatment, we anticipate that the effect of treatments received would not significantly affect the findings of our study. Lastly, the heterogeneous disease duration in our cohort suggests that use of Cox regression would be more appropriate as it entails time-to-event analyses. The lack of time-to-event data in our prevalent cohort precluded the use of Cox regression; nevertheless, we tried to overcome the potential bias of logistic regression, by performing subgroup analyses in patients with short disease duration.
In summary, despite recent advances, we found that almost 50% of patients with lupus initially presenting with mild disease eventually progress to more severe forms of the disease, highlighting the existence of persistent unmet needs in SLE. Milder forms of lupus may still carry an increased risk to ‘convert’ over time; thus, increased vigilance and regular monitoring are warranted in patients, irrespective of phenotype at disease onset.
Acknowledgments
We thank the staff physicians (Drs T Karageorgas, D Tseronis, M Aggelakos, D Kassara, K Thomas, E Atsali, S Boiu, L Fotis) and nurses (G Rapsomaniki, A Ntourou, K Togia, T Gerogianni) of the Rheumatology Unit of the 'Attikon' University Hospital of Athens for their referral and care to the patients with SLE. We are also indebted to Dr G Bertsias for his insightful comments.
References
Footnotes
DSN and MK contributed equally.
DTB and AF contributed equally.
Collaborators George Bertsias.
Contributors DSN and MK collected data from patient medical charts and also performed the statistical analysis. DSN performed the data entry. AP, SF, KC and AB contributed to maintenance of the Attikon Lupus Registry and assisted in data collection. PK contributed to establishment of the Attikon Lupus Registry. PK and JB assisted in patient recruitment and reviewed the manuscript. DTB and AF conceived and supervised the study. AF performed the statistical analyses. DSN and AF drafted the manuscript.
Funding This work was funded in part by the Hellenic Society of Rheumatology; the Foundation for Research in Rheumatology (FOREUM); the Greek General Secretariat for Research and Technology 'Aristeia' action of the Operational Programme 'Education and Lifelong Learning' (co-funded by the European Social Fund and National Resources, Aristeia I 2344 to DTB); the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 742390); and the SYSCID (A Systems Medicine Approach to Chronic Inflammatory Diseases) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 733100).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval The study was approved by the Ethics Committee of the 'Attikon' University Hospital of Athens (protocol number 103/06-03-2014).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.