Article Text

Abstract
SLE is a complex autoimmune disease with genetic, epigenetic, immune-regulatory, environmental and hormonal factors. Kidney inflammation and injury, termed lupus nephritis (LN), occurs in over half of patients with SLE and is a leading cause of disability and death. There is a high degree of short-term and long-term side effects associated with current LN therapies and they are not effective for many patients. Thus, novel therapies with reduced toxicity and improved efficacy are drastically needed. Many of the known LN susceptibility genes have functions that mediate inflammation via cytokine/chemokine production and activation of myeloid and B cells. Understanding the cellular and molecular mechanisms mediated by these variant gene products provides valuable insight for the development of improved and personalised diagnostics and therapeutics. This review describes variants in the TNIP1 (tumour necrosis factor α-induced protein 3-interacting protein 1) gene associated with risks for SLE and LN and potential roles for loss of function of its protein product ABIN1 in the activation of myeloid and B-cell-mediated injury in LN.
- lupus nephritis
- lupus erythematosus
- systemic
- polymorphism
- genetic
- inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
SLE is a worldwide autoimmune disease, with incidence and prevalence highest in North American at 23/100,000 person-years and 241/100,000 people, respectively.1 Patients are more likely to be female in childbearing years and people of Black Eethnicity have the highest incidence and prevalence.1 2 Lupus nephritis (LN) occurs at some point during the course of disease in 50%–60% of patients.3 4 LN is the most severe complication of SLE, resulting in increased morbidity and mortality. Current therapy produces a remission of LN in only 10%–40% by 12 months, and up to 30% of patients eventually develop renal failure requiring dialysis.5 6 Even patients who achieve a complete or partial remission with therapy average one recurrence of LN yearly. One of the best predictors of a good renal function outcome is remission of proteinuria within 6 months of initiating therapy. Those data suggest that rapid control of glomerular inflammation, as demonstrated by remission of proteinuria, is the primary goal of induction therapy. Initial treatment of LN uses high-dose corticosteroids to rapidly suppress leucocyte-mediated inflammatory activity and a second immunosuppressive drug to impair B-cell production of autoantibodies, a process that can require months of therapy.5 7 8 This treatment regimen markedly improves the outcome of what was a fatal disease, when untreated. Although improvements in immunosuppressive regimens have occurred, borrowing from oncology and transplant protocols, treatment to rapidly inhibit inflammation has not changed since the introduction of corticosteroids in the 1960s. Thus, defining novel inflammatory mediators in LN will provide important insight for improved therapeutics. The transcription factor NF-κB is a key regulator of proinflammatory cytokines and chemokines.9 Genetic studies have identified variants in several NF-κB regulatory genes as risks for SLE and LN.10 11 One such gene is tumour necrosis factor α (TNF-α)-induced protein 3-interacting protein 1 (TNIP1). TNIP1 expresses the protein A20 binding inhibitor of NF-κB (ABIN1) that functions as a physiological inhibitor of NF-κB by binding to polyubiquitin moieties on upstream regulators.12–15 There is an accumulation of evidence that suggests that TNIP1 mutation and subsequent loss of ABIN1 function contributes to the development of LN via enhanced activation of NF-κB. This review describes the molecular and cellular events that are activated by loss of ABIN1 function in LN and discusses potential clinical applications from this information.
TNIP1 variants show increased risks for SLE and LN
There is substantial evidence linking TNIP1 single-nucleotide polymorphisms (SNPs) to increased risk of developing SLE and LN (table 1). The most reported TNIP1 variant associated with SLE and LN is rs7708392. This SNP has been replicated in several different SLE cohorts including European and Chinese,16 17 North American,18 Egyptian,19 Japanese,20 Swedish21 and Hispanic22 populations. Our group found a strong association for rs7708392 with LN in European Americans (EA) in a large-scale study of North American patients with SLE.23 This study included 2161 EA SLE patients without LN, 1129 with LN and 3491 healthy EA individuals. Kawasaki et al report a significant association for rs7708392 in SLE patients with LN using a case–control association study in a Japanese cohort that included 364 patients with SLE and 553 patients with rheumatoid arthritis and 513 healthy individuals.20 A study of a Egyptian cohort that included 53 SLE patients with LN, 57 SLE patients without nephritis and 85 healthy controls showed that rs7708392 was significantly more prevalent among patients with LN than in patients without LN.19 However, there are discrepancies as to which allele of rs7708392 is associated with increased risk. Our group identified the minor C allele to be associated with increased risk for LN in North American patients with SLE with European ancestry. The C allele was also the minor risk allele identified in Japanese patients with LN.20 Conversely, Rizk et al determined that Egyptian patients with SLE had a 3.4-fold increase in risk of developing LN if they contained one G allele.19 Subsequently, Zhong et al found the G allele to be protective in the Chinese population, but given its low frequency in Asian populations (30%) compared with European populations (70%) they suggest there is genetic heterogeneity in this locus between different populations, which may explain the observed discrepancies.17
TNIP1 SNPs associated with SLE and LN
Several additional TNIP1 SNPs are associated with increased risk for SLE. Bolin et al found the rs6889239 SNP to be strongly associated with SLE in Swedish Caucasian patients.21 Adrianto et al found significant association with SLE risk with the SNP rs6889239 in American patients with European ancestry and the SNP rs13168551 in African-Americans (AA).22 Han et al found rs10036748 to be highly associated with SLE in Chinese populations.24 Associations with LN for those SNPs were not investigated and thus are unknown. However, we reported that the TNIP1 rs4958881 SNP has a very strong association with LN in AA patients with SLE in a study, which included 1811 healthy control AA, 709 AA SLE patients with LN and 704 AA SLE patients without LN.23 We recently genotyped an independent cohort of 230 European ancestry Americans and 225 AA patients with LN for the TNIP1 variant rs4958881 and 33% of the European ancestry Americans and 73% of the AA patients with LN have the rs4958881 risk allele (unpublished data). The risk allele was also significantly associated with class III, IV and V LN and crescent formation in African patients with LN with the rs4958881 SNP, but not for European ancestry patients with LN.
In summary, multiple large-scale and replicate genetic studies have identified TNIP1 variants as risk factors for SLE, of which the rs7708392 and rs4958881 polymorphisms display a significant increased risk of LN in various SLE cohorts (table 1 and figure 1). The causative effects of these SNPs in SLE or LN have not yet been reported. However, Adrianto et al identified two independent risk haplotypes (H1 and H2) with SNPs near the TNIP1 promoter and showed lower levels of TNIP1 mRNA and ABIN1 protein in EVB-transformed B-cell lines derived from European ancestry patients with SLE homozygous for H1 and H2.22 The H2 haplotype also contains a coding missense variant rs2233290 that would result in an ABIN1 P151A mutation. This residue is in a proline-rich region that has been to shown to facilitate protein folding and confirmation.25 Thus, the P151A mutation could disrupt the functional structure of ABIN1. Assessment of ABIN1 expression and activity in kidneys and circulating immune cells in patients with LN with different TNIP1 SNPs and target populations could provide important insight regarding the impact of TNIP1 variants on ABIN1 function in LN. The location/region for each identified TNIP1 polymorphism is shown in figure 1.

This diagram shows the location/region for each identified TNIP1 (tumour necrosis factor-α-induced protein 3-interacting protein 1) polymorphism listed in table 1 and colour code for the ethnicity of the cohort for which they were identified.
Effector cells and cellular and molecular events mediated by loss of ABIN1 function in LN
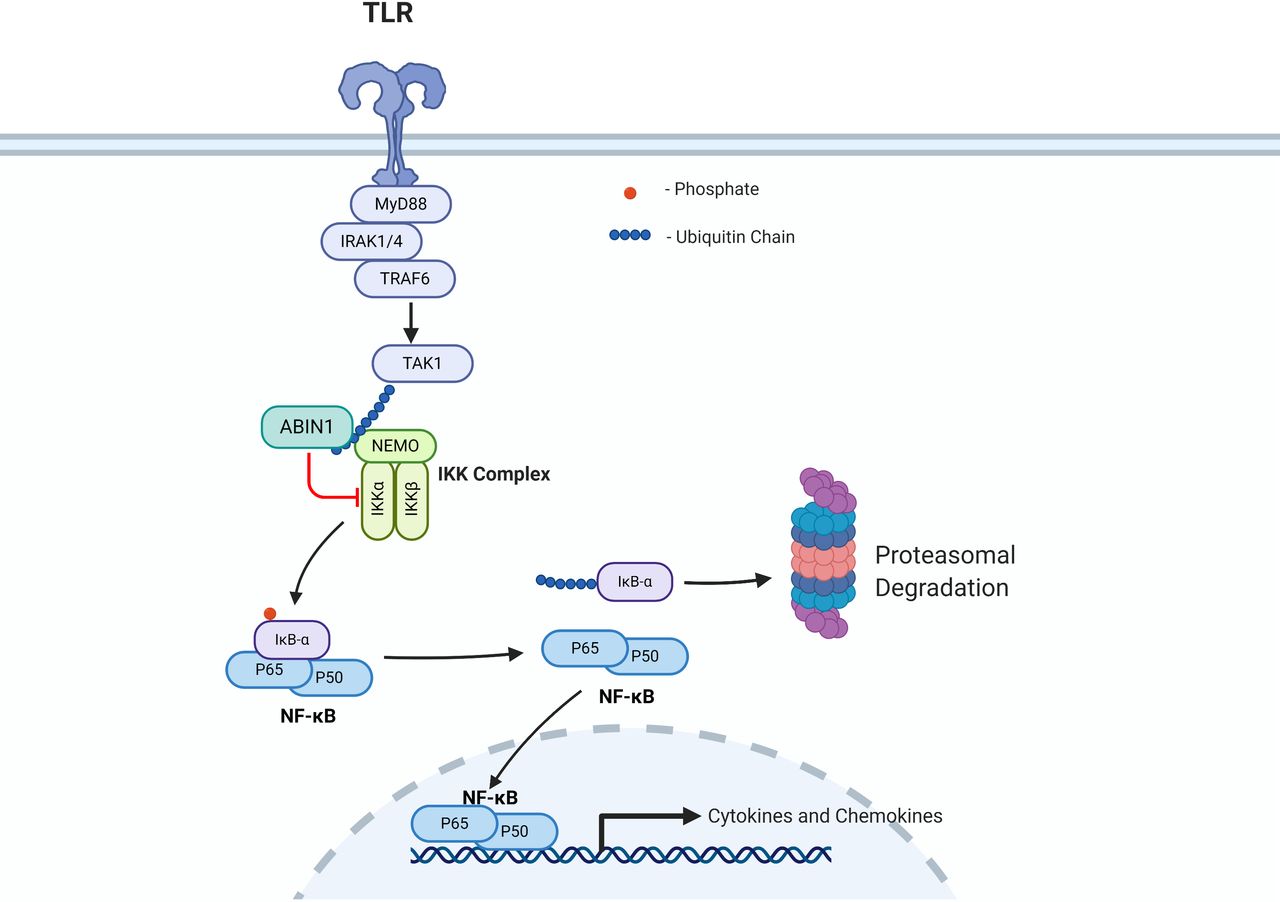
Although the direct causative effects of identified TNIP1 polymorphisms have not been determined, studies in mice indicate a role for loss of function of the TNIP1 product ABIN1 in the development of SLE and LN. There are also possible cellular mechanisms indicated by the loss of function of ABIN1. ABIN1 functions as a physiological inhibitor of NF-κB by binding to polyubiquitin moieties on key upstream regulators (figure 2) (also reviewed in Ref. 13). Molecular studies have demonstrated that ABIN1 regulates the canonical NF-κB pathway via binding to polyubiquitin moieties on upstream regulators and studies with ABIN1 knockout mice and ABIN1[D485N] mice (lacking ABIN1 polyubiquitin binding) have indicated dependence of different components of this signalling pathway in the development of LN.13 26–29 The canonical NF-κB pathway and regulation by ABIN1 are depicted in figure 2. NF-κB remains in the cytoplasm in an inactive state by binding with the inhibitor of κB (IκB)-α until receptor-dependent activation of IκB kinase (IKK) complex mediates phosphorylation of IκB-α. This phosphorylation event results in IκB-α release from NF-κB and its degradation. This allows NF-κB to translocate into the nucleus and activate transcription of hundreds of immune-modulating cytokines and chemokines.30 ABIN1 inhibits NF-κB transcriptional activation by binding to the essential component of the IKK complex (NEMO) and preventing phosphorylation of IκB-α and its subsequent degradation.13 Thus, a postulate is that TNIP1 variants result in enhanced activation of NF-κB that provokes immune and inflammatory cell-mediated injury characteristic of LN. Our group has reported that transgenic mice globally lacking the ubiquitin-binding activity and NF-κB inhibitory function of ABIN1 (ABIN1[D485N] mice) spontaneously and progressively develop a number of SLE-associated phenotypes, including splenomegaly, elevated serum level of pathogenic ANA and anti-double-stranded DNA (dsDNA) autoantibodies, as well as progressive glomerulonephritis (GN) with pathological features of human class IV LN.23 31 A recent report by Kuriakose et al showed that global TNIP1/ABIN1 deficient mice also spontaneously develop SLE-associated autoimmune phenotypes and progressive, proliferative immune complex-mediated GN.29

This is a simplified version of the canonical nuclear factor kappa B cell (NF-κB) pathway that is negatively regulated by A20 binding inhibitor of NF-κB (ABIN1) function. Other reviews describe NF-kB signalling and ABIN1 interactions and NF-κB regulation in greater detail.9 13 In brief, ABIN1 negatively regulates NF-kB by binding to polyubiquitin moieties on (TAK1, NEMO) inhibiting phosphorylation and degradation of inhibitor of κB-α (IkB-α), preventing nuclear translocation of NF-kB and inhibiting transcription of target immune regulators. Created with Biorender.com. IKK, IκB kinase; TLR, Toll-like receptor.
Role of inflammatory cells
Our group reported that glomerular immune cell recruitment and retention was enhanced in ABIN1[D485N] mice.23 32 Kuriakose et al further characterised this effect through marker expression analysis for tissue macrophage, classic inflammatory monocytes (iMo), patrolling monocytes (pMo) and neutrophils. Their analysis showed that glomerular pMo and neutrophil accumulation was significantly enhanced in ABIN1-deficient mice compared with wild-type mice.29 Interestingly, they report that mice with selective monocyte-targeted ABIN1 deficiency developed GN and had significant increases in glomerular pMo and neutrophil accumulation. Additionally, they crossed Nr4a1-deficient mice with TNIP1-deficient mice where the offspring have a genetic deletion of pMo, but still have ABIN1 knocked out in all other cells. These animals did not develop GN despite global loss of ABIN1, further supporting a critical role for loss of ABIN1 function in pMo in the development of LN. Studies assessing targeted knockout of ABIN1 in neutrophils were not included in the report. Nanda et al performed a similar flow cytometric-based profile for myeloid cell markers and found that the numbers of pMo and iMo were increased in blood and kidneys of ABIN1[D485N] mice compared with wild-type control mice.27 Collectively, these findings suggest that loss of ABIN1 function in iMo, pMo and neutrophils contributes to their production, glomerular influx and glomerular injury in LN.
Role of adaptive immune cells
Production of ANA, anti-dsDNA autoantibodies and deposition of immune complexes within the glomerulus are well-known pathological indications of SLE and LN.33 Plasma B cells produce antibodies and production of autoantibodies from germinal centre B (GCB) cells can trigger the development of LN.27 34 IL-6 stimulation of splenic GCB cells is required for anti-dsDNA and ANA production.35 ABIN1[D485N] mice crossed to IL-6 knockout mice had reduced splenomegaly and abolished GCB cell formation, reduced serum anti-dsDNA, reduced total IgM, IgG and IgE and suppressed GN development.27 RAG-2 deficient mice lack mature B and T cells and cannot develop GCB cells or autoantibodies.36 Crossing of RAG-2 deficient mice with ABIN1[D485N] mice also completely suppressed development of GN.27 In contrast, Kuriakose et al found that crossing Rag-1 deficient mice, which also lack mature T and B cells to ABIN1 knockout mice still develop GN.29 This difference is unclear, but might be related to loss of additional functions of the ABIN1 protein in the ABIN1-deficient mice, versus specific loss of ubiquitin-binding function due to the ABIN1[D485N] mutation. These reports suggest that loss of ABIN1 ubiquitin-binding function in adaptive immune cells contributes to autoantibody production and glomerular immune complex deposition in SLE.
Role of intrinsic kidney cells
To investigate a role for intrinsic kidney cells in ABIN1-mediated GN, our group used exogenous nephrotoxic serum (NTS) injections to induce acute immune-complex GN in ABIN1[D485N] and wild-type mice.32 These experiments were performed at an age prior to spontaneous development of disease phenotypes in ABIN1[D485N] mice. NTS-mediated nephritis is a well-characterised model of acute LN in which glomerular neutrophil accumulation occurs within 2 hours and proteinuria occurs at 24 hours in wild-type mice.37 NTS-induced glomerular neutrophil recruitment and retention and proteinuria were significantly enhanced in ABIN1[D485N] mice.32 Cultured human-derived podocytes with ABIN1[D472N] mutation (human D485N homologue) were used to investigate a role for podocytes in this process.32 Transcription and secretion of several TNF-α-stimulated NF-κB-target cytokines and chemokines were greatly enhanced in ABIN1[D472N] podocytes and supernatants from these cells activated neutrophil migration. ABIN1[D472N] podocytes were also more prone to cytoskeletal rearrangement and subsequent morphological disruption following incubation with neutrophil granule products. This type of podocyte morphological change is indicative of podocyte injury that results in glomerular filtration disruption in GN.38 Administration of an inhibitor of neutrophil granule release also attenuated NTS-induced podocyte injury and proteinuria in ABIN1[D485N] mice.32 These results suggest that there is interplay between podocytes and neutrophils in glomerular inflammation and injury in LN that is mediated by loss of ABIN1 ubiquitin-binding function in these cells.
Other studies indicate specific molecular events that contribute to ABIN1-mediated LN. Nanda et al showed that the ABIN1[D485N] mutation disrupts binding to polyubiquitin chains and polyubiquitinated NF-κB regulatory proteins in vitro.28 Naive splenic B cells and bone marrow-derived myeloid cells isolated from ABIN1[D485N] mice also displayed enhanced Toll-like receptor (TLR) 2/6, 4 and 7-mediated phosphorylation of the canonical IKKs and IκB-α proteins and production of proinflammatory cytokines.28 MyD88, IRAK1 and IRAK4 are part of the signalling cascade used by TLR7 and TLR9 to activate the IKK complex.39 Nanda et al reported that crossing ABIN1[D485N] mice with TLR7- and MyD88-deficient mice or mice with IRAK1- or IRAK4-inactivating mutations suppressed development of autoimmunity and GN while also diminishing pMo and iMo increases in blood.26–28 Administration of an IRAK4-specific inhibitor also reduced blood pMo number and attenuated GN in ABIN1[D485N] mice and reduced TLR7 agonist-stimulated cytokine secretions from pMo and iMo in vitro.27 Kuriakose et al found that knocking out MyD88 and combination of TLR7 and TLR9 reduced glomerular accumulation of pMo and neutrophils and prevented the development of GN in ABIN1-deficient mice.29 Collectively, these results indicate that the contribution of ABIN1 loss of function in innate and adaptive immune cells to LN is mediated by enhanced activation of the canonical NF-κB pathway in these cells.
Role for loss of ABIN1 function in elevated cytokine and chemokine levels in LN
High urine and serum concentrations for numerous NF-κB-regulated proteins have been implicated as biomarkers in LN (extensively reviewed)40 41, but the mechanisms for these increased levels are unknown. Perhaps, the most-reported and validated urinary biomarkers of this type are the chemokines monocyte chemoattractant protein-1 (MCP-1) and interferon gamma-induced protein 10 (IP-10). A recent meta-analysis that included seven studies for IP-10 and another that included eight studies for MCP-1 found that urinary, but not serum levels of MCP-1 and IP-10 were significantly elevated in patients with active versus inactive LN.42 43 Several reports have demonstrated that higher serum levels of interleukin (IL)-6, IL-17 and TNF-α are associated with active LN.44–47 It is possible that that elevated urine and serum levels of these inflammatory activators in LN is mediated by enhanced expression in immune cells via loss of ABIN1 function (figure 2). Enhanced expression and secretion from intrinsic kidney cells could also contribute to elevated urine levels. Our group performed custom qRT-PCR analysis for NF-κB-target genes with isolated glomeruli from ABIN1[D485N] and wild-type mice following induction of immune complex GN with NTS.32 NTS induced a twofold increase in IL-17 and MCP-1 and sixfold increase in TNF-α expression in glomeruli from ABIN1[D485N] compared with wild-type mice. We performed a similar analysis with human-derived podocytes with native ABIN1 or ABIN1[D472N] (D485N homologue) stimulated with TNF-α and found that MCP1, IL-8 and TNF-α mRNA expression was enhanced in ABIN1[D472N] podocytes. ELISA results showed that TNF-α-induced secretion of MCP1, IL-8 and TNF-α was also enhanced in ABIN1[D472N] cells. In addition, IP-10 mRNA expression was 10-fold higher in ABIN1[D472N] podocytes than wild-type cells following TNF-α stimulation. We found that kidney, plasma and urine IP-10 concentrations were also significantly higher in ABIN1[D485N] versus wild-type mice, at an age where severe proliferative GN occurs in ABIN1[D485N] mice (unpublished data). Nanda et al reported that TLR-stimulated TNF-α and IL-6 secretion were enhanced in bone marrow-derived myeloid cells and TLR-stimulated IL-6 was enhanced in B cells isolated from ABIN1[D485N] mice versus wild-type mice.28 This group showed in a another report that TLR7 activation of TNF-α and IL-6 was also enhanced in pMo and iMo isolated from ABIN1[D485N] mice.27 These findings indicate that TNIP1 mutation and subsequent loss of ABIN1 function in immune and kidney cells contribute to elevated urinary or serum levels of MCP-1, IP-10, Il-8, IL-17, TNF-α and IL-6 in LN.
Clinical applications
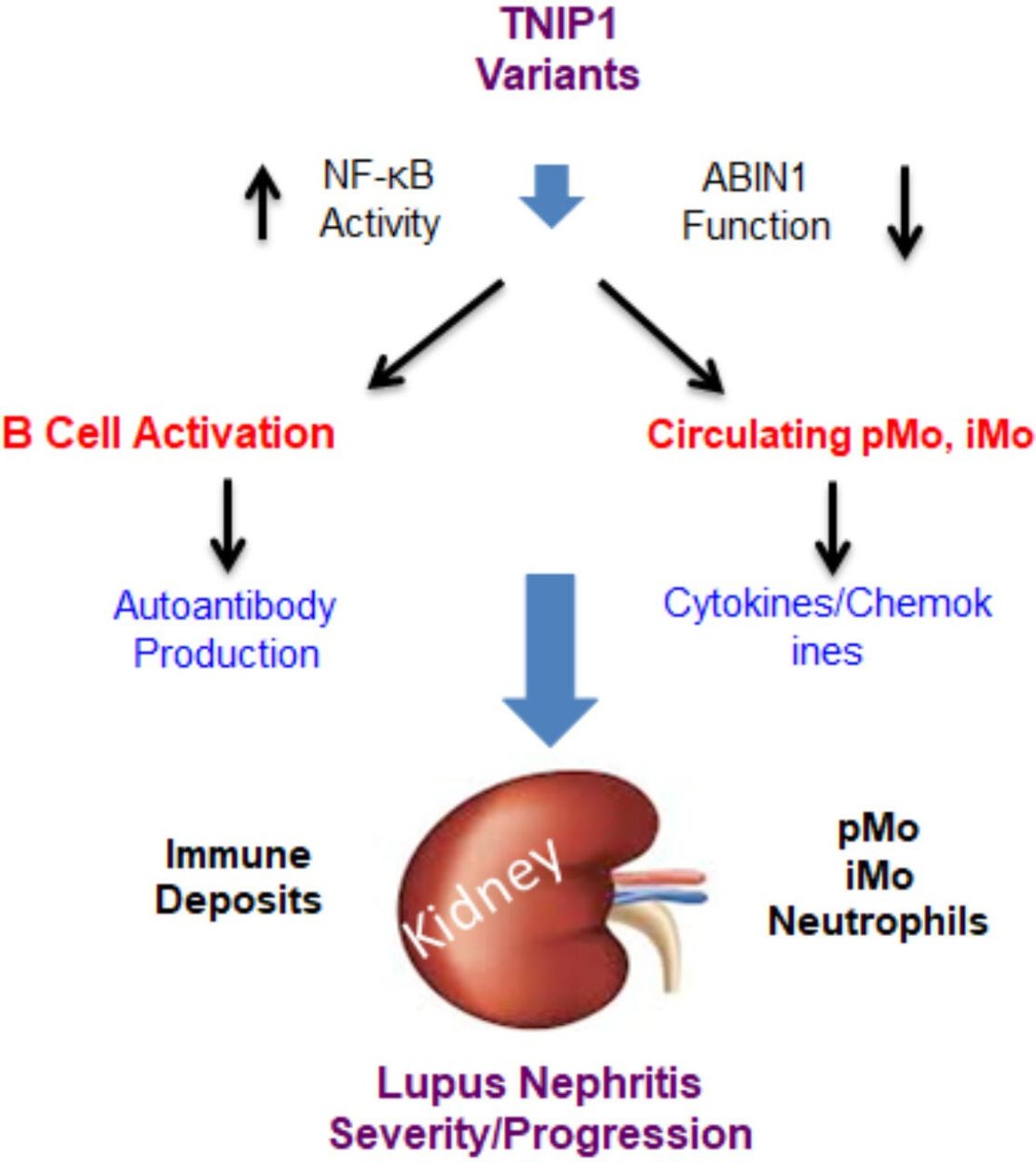
Multiple TNIP1 variants contribute to risks of SLE and LN development.16–24 Animal models and in vitro studies demonstrate that loss of ABIN1 function leads to stimulation of NF-κB-mediated cytokine and chemokine production in target cells and enhanced production and activations of myeloid and B cells in LN.27 28 32 Together, these findings support an overall model for TNIP1 mutations in the development of LN (figure 3). TNIP1 rs7708392 and rs4958881 variants may help identify patients at increased risk of developing LN, especially in populations for which they were identified to increase risk. TNIP1 variants identified as risks for SLE (rs6889239, rs10036748, rs13168551), but not investigated in LN should also be assessed in patients with LN. These findings also provide important insight into novel therapeutic targets and provide an opportunity to use personalised medicine for those with TNIP1 variants. Potential molecular targets include inhibition of IRAK4, MyD88 and TLR 7/9 and their downstream targets. This includes the use of monoclonal antibodies to neutralise NF-κB-regulated cytokines that are implicated in human LN and activated by loss of ABIN1 function in mice.27 28 32 40 41 Monoclonal antibodies for IL-6 (sirukumab), IL-23/17 (ustekinumab) and IFN-γ blockade (AMG 811) are currently available or being tested in patients with LN.41 48–52 Another potential NF-κB-targeted chemokine is CXCL10 (C-X-C motif chemokine ligand 10)/γ-interferon IP-10. Studies have shown that serum levels of IP-10 are increased in patients with class IV proliferative LN and phase II trials for ulcerative colitis demonstrate safety and effectiveness of monoclonal IP-10 antibody (eldelumab) treatment.53–55 In conclusion, a combination of TNIP1 variant genotyping and serologic and urine analysis for downstream NF-κB-mediated cytokines and chemokines will possibly provide precision diagnostics and personalised therapeutics for LN.

{kind=link}
{kind=link}
{kind=link}
This represents a proposed model for how genetic variants for the A20 binding inhibitor of NF-κB (ABIN1) gene TNIP1 (tumour necrosis factor-α-induced protein 3-interacting protein 1) contribute to the development and progression of SLE and lupus nephritis (LN). A number of TNIP1 single-nucleotidepolymorphisms have been identified in association with SLE and LN from large-scale genome-wide association study and replicate studies.16–24 ABIN1 is ubiquitously expressed. It is possible that TNIP1 variants result in reduction or loss of ABIN1 cellular function and enhanced activation of nuclear factor kappa B cell (NF-κB). The findings presented herein suggest that loss of ABIN1 function mediates activation of adaptive immune and myeloid cells resulting increased circulating monocytes and elevated blood autoantibody and inflammatory modulator levels. This subsequently results in increased glomerular leucocyte infiltration and immune deposition in LN. iMo, inflammatory monocyte;pMo, patrolling monocyte.
Future directions
There are important next steps in further defining the clinical impact of TNIP1 variant and ABIN1 dysfunction in LN. A recent report used a single-cell RNA sequencing (RNASeq) approach to profile and analyse gene expression in immune cells isolated from LN patient kidney biopsies.56 Twenty-two different immune cell clusters or subtypes were identified and the expression of 180 genes previously identified from SLE GWAS was assessed. Fifty-two of these genes were presented that demonstrated high variability across the 22 cell clusters, but TNIP1 was not included using these criteria. There are no other reports for TNIP1 or ABIN1 expression in LN. Thus, it is important to assess and compare TNIP1 and ABIN1 expression and activity in intrinsic kidney cells (podocytes, mesangial cells, endothelial cells) and kidney infiltrating and circulating immune cells in patients with LN with different TNIP1 variants versus patients with LN without the risk alleles. Assessing the immune cell RNASeq data in context of LN patient with TNIP1 variants compared with without would also provide valuable insight into the molecular events mediated by ABIN1 dysfunction in LN. Lastly, a specific role for ABIN1 dysfunction in LN could be determined by TNIP1 genotyping and ABIN1 expression analyses in kidneys and immune cells from patients with other types of nephritis (IgG nephropathy, membranous nephropathy, etc).
References
Footnotes
Contributors MPB and DWP contributed to the initial conception and organisation of this review article. All authors contributed to the composition and editing.
Funding DJC was supported by a National Institute of Diabetes and Digestive and Kidney Diseases K08 Career Development Award DK102542.
Competing interests DJC reports personal fees from Retrophin, GSK and Aurinia.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.