Article Text

Abstract
Objective Recently, podocytes have been recognised not only as a physical barrier to prevent urinary protein loss but also as producers of proinflammatory cytokines. However, the roles of podocytes in the pathogenesis of lupus nephritis (LN) remain largely unknown. This study aims to determine the roles of suppressor of cytokine signalling (SOCS) family members expressed in glomeruli in the regulation of LN.
Methods We investigated the expression of SOCS family members in glomeruli in murine lupus model induced by repeated epicutaneous administration of the TLR7/8 agonist imiquimod. We also investigated the roles of SOCS3 expressed in podocytes in the imiquimod-induced glomerulonephritis and systemic autoimmunity by using podocyte-specific SOCS3-deficient mice (podocin-Cre x SOCS3fl/fl mice (SOCS3-cKO mice)). Finally, we investigated the expression of proinflammatory cytokines and chemokines in SOCS3-deficient podocyte cell lines.
Results qPCR analysis revealed that among SOCS family members, SOCS3 was preferentially induced in glomeruli on epicutaneous administration of imiquimod and that interleukin 6 (IL-6) induced SOCS3 expression in podocyte cell lines. SOCS3-cKO mice exhibited severe glomerulonephritis, high levels of serum creatinine and urine albumin and decreased survival rate compared with control SOCS3-WT mice. Levels of anti-double-strand DNA antibody, SOCS (GC) formation and the numbers of follicular helper T (Tfh) cells and GC B cells in the spleen were higher in SOCS3-cKO mice than those in SOCS3-WT mice. Serum IL-6 levels and expression of IL-6 mRNA in glomeruli were also elevated in SOCS3-cKO mice. IL-6-induced IL-6 expression was enhanced in SOCS3-deficient podocyte cell lines compared with that in SOCS3-sufficient podocyte cell lines.
Conclusion SOCS3 expressed in podocytes plays protective roles for the development of glomerulonephritis and inhibits autoantibody production in the imiquimod-induced lupus model presumably by suppressing IL-6 production of podocytes.
- autoimmune diseases
- cytokines
- lupus erythematosus
- systemic
Data availability statement
Data are available in a public, open access repository. Kotaro Suzukisuzuki_k@faculty.chiba-u.jp.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Recently, podocytes have been recognised not only as a physical barrier to prevent urinary protein loss but also as producers of proinflammatory cytokines.
What does this study add?
Suppressor of cytokine signalling 3 (SOCS3) was preferentially induced in glomeruli in murine lupus model induced by imiquimod and podocyte-specific SOCS3-deficient mice (SOCS3-cKO mice) exhibited severe glomerulonephritis, high levels of serum creatinine and urine albumin compared with control mice.
Levels of anti-double-strand DNA antibody, germinal centre (GC) formation and the numbers of follicular helper T cells and GC B cells in the spleen were higher in SOCS3-cKO mice than those in control mice.
Serum interleukin 6 (IL-6) levels and expression of IL-6 mRNA in glomeruli were elevated in SOCS3-cKO mice.
How might this impact on clinical practice or future developments?
Our data suggest that the induction of SOCS3 in podocytes could be a therapeutic approach for treating SLE via the blockade of IL-6 signalling.
Introduction
SLE is a representative systemic autoimmune disease that shows various damage in multiple organs. Especially, lupus nephritis (LN) is one of the most common manifestations in SLE, and LN leads to end-stage kidney disease if nephritis is uncontrolled. Indeed, despite intensive therapies, patients with SLE with LN suffer from increased mortality compared with those without LN.1 It is considered that autoantibodies such as anti-double-strand DNA (anti-dsDNA) antibodies cause inflammation and damage in the kidney, leading to the development of LN.2 In addition, many cytokines are known to be involved directly or indirectly (via the production of autoantibodies) in the development of LN.
Many cytokines utilise Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathways for transducing their signals.3 Consequently, JAK-STAT pathways play multiple roles in cell differentiation, proliferation, activation and survival in many cell types3 and are involved in the pathogenesis of various autoimmune diseases including rheumatoid arthritis.4 In addition, a recent study has shown that the JAK-STAT pathways are involved in the pathogenesis of SLE in humans.5 Moreover, accumulating evidence suggests that suppressor of cytokine signalling (SOCS) family proteins, representative cytokine-inducible negative regulators of JAK-STAT pathways, plays crucial roles in the regulation of inflammation and autoimmune responses.3 However, the roles of SOCS family members in the pathogenesis of LN remain unclear.
Podocytes are highly specialised, terminally differentiated epithelial cells, which function as a physical barrier to prevent urinary protein loss through the special formation and maintenance of the foot process. In addition to the barrier function, a recent study has shown that podocytes produce various cytokines such as tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6), interleukin 8 (IL-8), vascular endothelial growth factor (VEGF), granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF),6 suggesting that podocytes could contribute to the inflammatory process during glomerulonephritis. However, the precise roles of podocytes in the pathogenesis of LN are largely unknown.
In the present study, we address this point by using a murine lupus model with podocyte-specific conditional knockout mice. Our findings indicate that SOCS3 expressed in podocytes plays protective roles for the development of glomerulonephritis and inhibits autoantibody production in the lupus model possibly by suppressing IL-6 production of podocytes.
Methods
Mice
Podocin-Cre knock-in (podocin-Cre) mice7 and SOCS3fl/fl mice8 were backcrossed onto BALB/c mice (Japan SLC, Shizuoka, Japan) over 10 generations. Podocin-Cre mice were mated to SOCS3fl/fl mice to generate podocyte-specific SOCS3-deficient mice (podocin-Cre × SOCS3fl/fl mice; SOCS3-cKO mice). Littermate SOCS3fl/fl mice (SOCS3-WT mice) were used as controls. Lupus-prone MRL/MpJJmsSlc-lpr/lpr (MRL/lpr) mice and control MRL/MpJJmsSlc-+/+ (MRL/+) mice were purchased from Japan SLC. All mice were housed in micro isolator cages under specific pathogen-free conditions, and all experiments were performed according to the guidelines of Chiba University.
Isolation of murine glomeruli
Murine glomeruli were isolated as described previously.9 Briefly, mice were anaesthetised and perfused with 8×107 Dynabeads (Thermo Fisher Scientific, Waltham, Massachusetts, USA) diluted in 40 mL of phosphate-buffered saline through the heart. Kidneys were removed, minced into 1 mm3 pieces and digested in digestion solution (1 mg/mL collagenase A, 100 U/mL deoxyribonuclease I in Hanks balanced salt solution) at 37°C for 30 min with gentle agitation. The digested tissues were gently passed through a 100 µm cell strainer with 5 mL of HBSS and centrifuged at 200 × g for 5 min. After washing with HBSS, glomeruli containing Dynabeads were gathered by a magnetic particle concentrator. During the procedure, kidney tissues were kept at 4°C except for the collagenase digestion process.
In some experiments, single-cell suspensions were prepared from isolated glomeruli as described elsewhere.9 In brief, isolated glomeruli were incubated with 0.2% trypsin-EDTA, 100 µg/mL heparin and 100 U/mL DNase I in HBSS for 25 min at 37°C, with mixing by pipetting. After the cell suspension was sieved through a 30 µm pore size filter, cells were collected by centrifugation at 200 × g for 5 min.
Imiquimod-induced lupus model
Female mice (7–9 week-old) were epicutaneously treated with imiquimod to induce a lupus-like disease.10 In brief, the right ear was painted topically with either 1.25 mg of 5% imiquimod cream (Glenmark, Mahwah, New Jersey, USA) in 100 µL of acetone or acetone alone (as a negative control) three times per week. Glomerulonephritis was evaluated at 4, 8 or 12 weeks after the initiation of topical administration.
Quantitative real-time PCR analysis
Total RNA was isolated and reverse transcription was performed as described previously.11 Quantitative RT-PCR was performed with a standard protocol on ABI PRISM7300 instruments (Applied Biosystems, Foster City, California,USA) using a SYBR green reagent (Applied Biosystems). The levels of each gene were normalised to the levels of GAPDH. The sequences of PCR primers were described in online supplemental methods.
Supplemental material
Cell culture
The conditionally-immortalised murine podocyte cell line was developed by the transfection with the temperature-sensitive T-antigen as described previously12 and was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum at 33°C. To induce differentiation, cells were cultured at 37°C for more than 14 days. Cells were then stimulated with or without IL-6 (10 ng/mL, R&D Systems, Minneapolis, Minnesota, USA) for 2 hours.
CRISPR-Cas9-mediated SOCS3 deletion in a murine podocyte cell line
The clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) gene-editing system was applied to establish murine podocyte cell lines lacking SOCS3.13 In brief, a single guide RNA (sgRNA) was designed to target the mouse SOCS3 exon 2 using the http://crispor.tefor.net website (sgRNA sequence (5′−3′): GCTGCTCAGCGCCGAGCCCG). The sgRNA was inserted downstream of the U6 promoter in a pHL-H1-ccdB-EF1a-RiH plasmid. Murine podocyte cell lines were transfected with a pHL-EF1a-SphcCas9-iP-A plasmid (Addgene plasmid # 60599; http://n2t.net/addgene:60599; RRID:Addgene_60599) and the pHL-H1-ccdB-EF1a-RiH using Lipofectamine 3000 (Thermo Fisher Scientific). After cells were cultured with hygromycin and puromycin for 48 hours, a single cell was cultivated in each well of a 96-well plate by a limited dilution method. Deletion of the SOCS3 gene was confirmed by immunostaining with anti-SOCS3 antibodies.
Histological analysis
According to the standard protocol, the kidney was fixed in 10% buffered formalin and embedded in paraffin, and sections were stained with periodic acid-Schiff (PAS). Glomerular injury was scored on a semiquantitative scale ranging from 0 to 4+ (1+ : focal, mild or early proliferative; 2+ : multifocal proliferative with increased matrix and inflammatory cells; 3+ : diffuse proliferative; 4+ : extensive sclerosis /crescents) as described elsewhere.14 Global glomerular lesion scores were calculated for 50 glomeruli in each mouse.
Immunostaining
The kidney was embedded in an OCT compound (Miles Laboratories, Elkhart, Indiana, USA), and cryostat sections were immunostained with Fluor 488-conjugated goat anti-mouse IgM antibody (Invitrogen, Carlsbad, California,USA). Murine podocyte cell lines were immunostained with FITC-conjugated rabbit anti-mouse SOCS3 antibody (Biorbyt LLC, San Francisco, California,USA).
Serological and urine analyses
Levels of plasma creatinine were determined by an EIA kit (Cayman Chemical, Ann Arbor, Michigan, USA). Urine was collected by using a metabolic cage, and the levels of urine albumin were quantified by a bromocresol green method.15
Enzyme-linked immunosorbent assay
Amounts of IL-6 were determined by using an ELISA kit from R&D Systems. Levels of anti-dsDNA antibodies were determined by an ELISA kit from Shibayagi (Shibukawa, Japan).
FACS analysis
Cells were stained and analysed on the FACSCanto II (Becton Dickinson, San Jose, California,USA) by using FlowJo software (TreeStar, Ashland, Oregon, USA). The following antibodies were used: anti-CD4 (RM4-5; BioLegend, San Diego, California,USA), anti-CXCR5 (2G8, BD Biosciences, San Jose, California,USA), anti-PD-1 (J43, BD Biosciences), anti-B220 (RA3-6B2, BioLegend), anti-Fas (SA367H8, BioLegend) and anti-GL7 (GL7, BioLegend). Before staining, Fc receptors were blocked with anti-CD16/32 antibody (2.4G2, BioLegend). Negative controls consisted of isotype-matched, directly conjugated, non-specific antibodies (BD Biosciences). In some experiments, single-cell suspensions of isolated glomeruli were incubated with 2 µM monensin (Sigma-Aldrich) in RPMI 1640 medium for 5 hours, and intracellular staining of SOCS3 and IL-6 and staining of podocin (NPHS2) were performed using anti-SOCS3 antibody (OTI3D3; Abcam, Cambridge, Massachusetts, USA), anti-IL-6 antibody (MP5-20F3; BD Biosciences) and anti-NPHS2 antibody (JB51-33; Hubio, Toronto, Canada).
Isolation of murine CD4+ T cells, CD8+ T cells and B cells
Naive CD4+ T cells, naive CD8+ T cells and B cells were isolated from spleens by using a naive CD4+ T cell isolation kit, a naive CD8a+ T cell isolation kit and a B cell isolation kit (Militenyi Biotec, Auburn, California,USA), respectively.
Quantification of NP-specific IgG
Mice were immunised intraperitoneally with 4-hydroxy-. 3- nitrophenylacetyl (NP)-ovalbumin (OVA) emulsified with IFA (OVA+IFA; Difco, Detroit, Michigan, USA). Seven days later, sera were collected, and the levels of NP-specific IgG1 and IgG2a were measured by ELISA as described previously.16
Detection of GCs
The numbers and areas of germinal centres (GCs) in the spleen were evaluated as described elsewhere.17
Data analysis
Data are summarised as means±SEM. The statistical analysis of the results was performed by either the unpaired Student’s t test or analysis of variance followed by Dunnett’s test. The survival analysis was performed by the log-rank test. <0.05 was considered significant.
Results
SOCS3 is induced in glomerular podocytes in a murine lupus model
Accumulating evidence suggests that SOCS family proteins play crucial roles in the regulation of inflammatory responses by inhibiting the JAK-STAT signalling.3 To clarify the roles of SOCS family members in LN, we first analysed mRNA expression of SOCS family members in glomeruli in an imiquimod-induced lupus model in which mice were epicutaneously administered with the TLR7/8 agonist imiquimod three times per week.10 As shown in figure 1A, qPCR analyses showed that, among SOCS family members, SOCS3 was preferentially induced in glomeruli at 4 weeks after the imiquimod administration. Because IL-6 activates STAT3, a potent inducer of SOCS3,3 in podocytes,18 we next examined whether IL-6 induces SOCS3 expression in a conditionally immortalised murine podocyte cell line.12 Indeed, IL-6 induced SOCS3 (figure 1B) but not other SOCS family members (data not shown) in the podocyte cell line. To confirm SOCS3 expression in podocytes in vivo, we quantified SOCS3 mRNA in glomeruli isolated from mice lacking SOCS3 expression specifically in podocytes (podocin-Cre x SOCS3fl/fl mice (SOCS3-cKO mice)) and control SOCS3-sufficient mice (SOCS3fl/fl mice) (SOCS3-WT mice) in the imiquimod-induced lupus model. The levels of SOCS3 mRNA in glomeruli were significantly increased at 4 weeks after the administration of imiquimod in SOCS3-WT mice but not in SOCS3-cKO mice (figure 1C). These results suggest that podocyte is the major cell type that expresses SOCS3 in glomeruli in the imiquimod-induced lupus model.
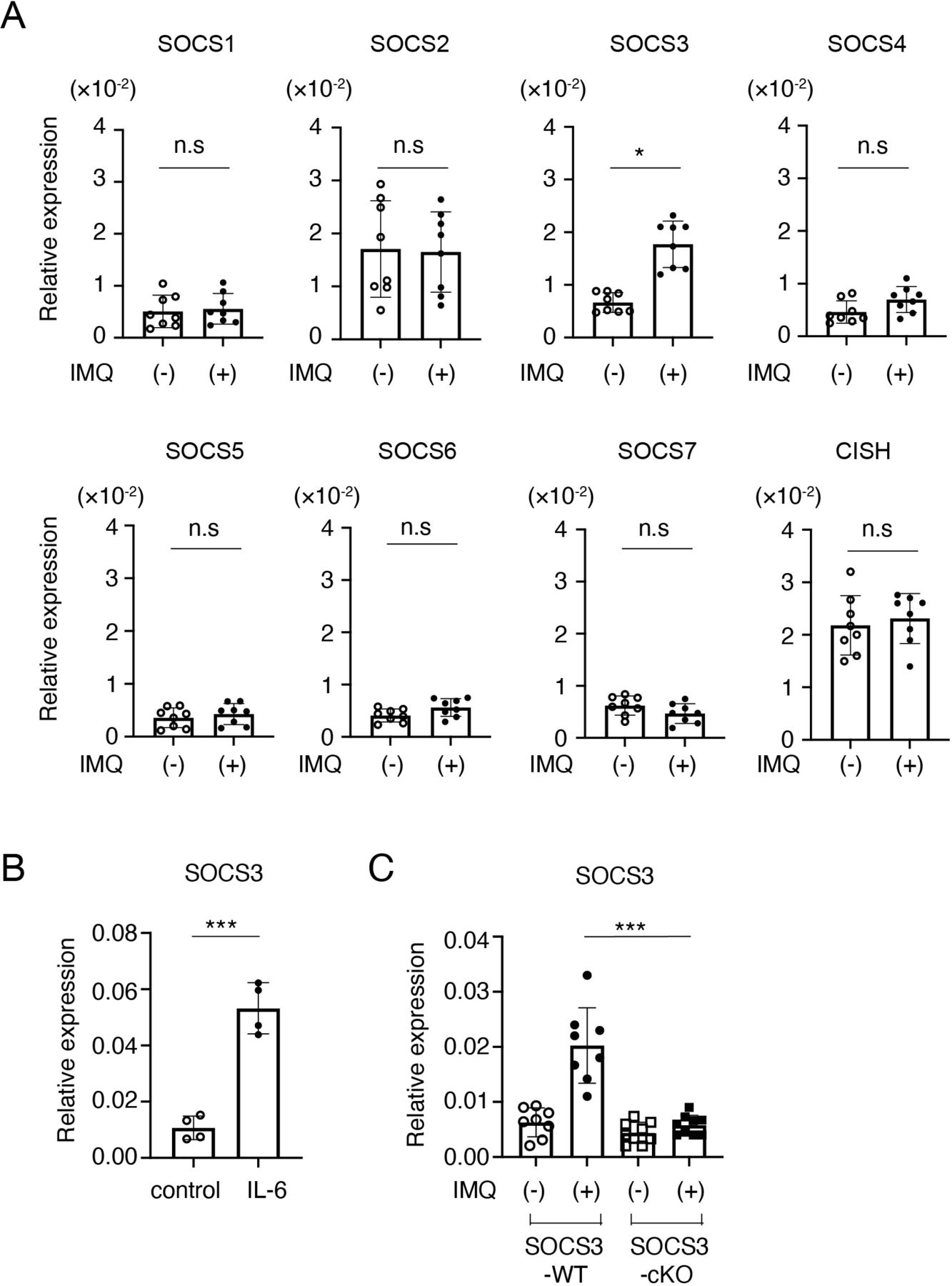
SOCS3 is induced in podocytes in the imiquimod-induced lupus model. (A) BALB/c mice were treated with or without topical imiquimod for 4 weeks. Glomeruli were isolated by the Dynabeads perfusion method. The expression levels of SOCS family members in the glomeruli were evaluated by qPCR analysis. Data are means±SEM for eight mice in each group. Data are compiled from two independent experiments. *p<0.05. (B) Conditionally immortalised murine podocytes were stimulated with or without IL-6 for 2 hours, and the expression levels of SOCS3 were evaluated by qPCR analysis. Data are compiled of four independent experiments. ***p<0.001. (C) Mice lacking SOCS3 expression in podocytes (podocin-Cre × SOCS3fl/fl mice (SOCS3-cKO mice)) and SOCS3-sufficient mice (SOCS3fl/fl mice) (SOCS3-WT mice) were treated with or without topical imiquimod for 4 weeks. Glomeruli were isolated and the expression levels of SOCS3 in the glomeruli were evaluated by qPCR analysis. Data are means±SEM for eight to nine mice in each group. Data are compiled from two independent experiments. ***p<0.001. CISH, cytokine-inducible SH2-containing protein; IMQ, imiquimod; KO, knockout; ns, not significant; SOCS, suppressor of cytokine signalling; WT, wild type.
SOCS3-cKO mice develop severe glomerulonephritis
To clarify the role of SOCS3 expressed in podocytes in LN, we next compared the severity of imiquimod-induced LN in SOCS3-cKO mice and control SOCS3-WT mice. First, we performed a histopathological examination of the kidney. We found that SOCS3-cKO mice developed more severe glomerulonephritis than SOCS3-WT mice did at 8 weeks after the topical administration of imiquimod (figure 2A,B). Consistent with the histopathological analyses, SOCS3-cKO mice showed elevated serum creatinine (figure 2C), increased urine albumin (figure 2D) and increased IgM deposition in glomeruli (figure 2E,F) compared with SOCS3-WT mice. These results suggest that SOCS3 expressed in podocytes attenuates glomerulonephritis in the imiquimod-induced lupus model.

SOCS3-cKO mice exhibit severe glomerulonephritis in the imiquimod-induced lupus model. SOCS3-cKO mice and SOCS3-WT mice were treated with or without topical imiquimod as described in the Methods section. (A) Representative photomicrograph of periodic acid-Schiff (PAS)-stained kidney sections from SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. (B) Renal histopathologic scores in SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. Data are means±SEM for 10 mice in each group. Data are compiled from two independent experiments. *p<0.05. (C) Serum creatinine levels in SOCS3-cKO mice and SOCS3-WT mice at 12 weeks. Data are means±SEM for 10–12 mice in each group. Data are compiled from two independent experiments. **p<0.01. (D) Urine albumin levels in SOCS3-cKO mice and SOCS3-WT mice at 12 weeks. Data are means±SEM for 8–9 mice in each group. Data are compiled from two independent experiments. *p<0.05. (E) Renal sections of SOCS3-cKO mice and SOCS3-WT mice at 8 weeks were stained with anti-IgM antibody. Shown are representative photographs. (F) The frequencies of the glomeruli having IgM deposits in 20 glomeruli were evaluated. Data are means±SEM for 10 mice in each group. Data are compiled from two independent experiments. ***p<0.001. IMQ, imiquimod; KO, knockout; SOCS, suppressor of cytokine signalling; WT, wild type.
Autoantibody production is accelerated in SOCS3-cKO mice
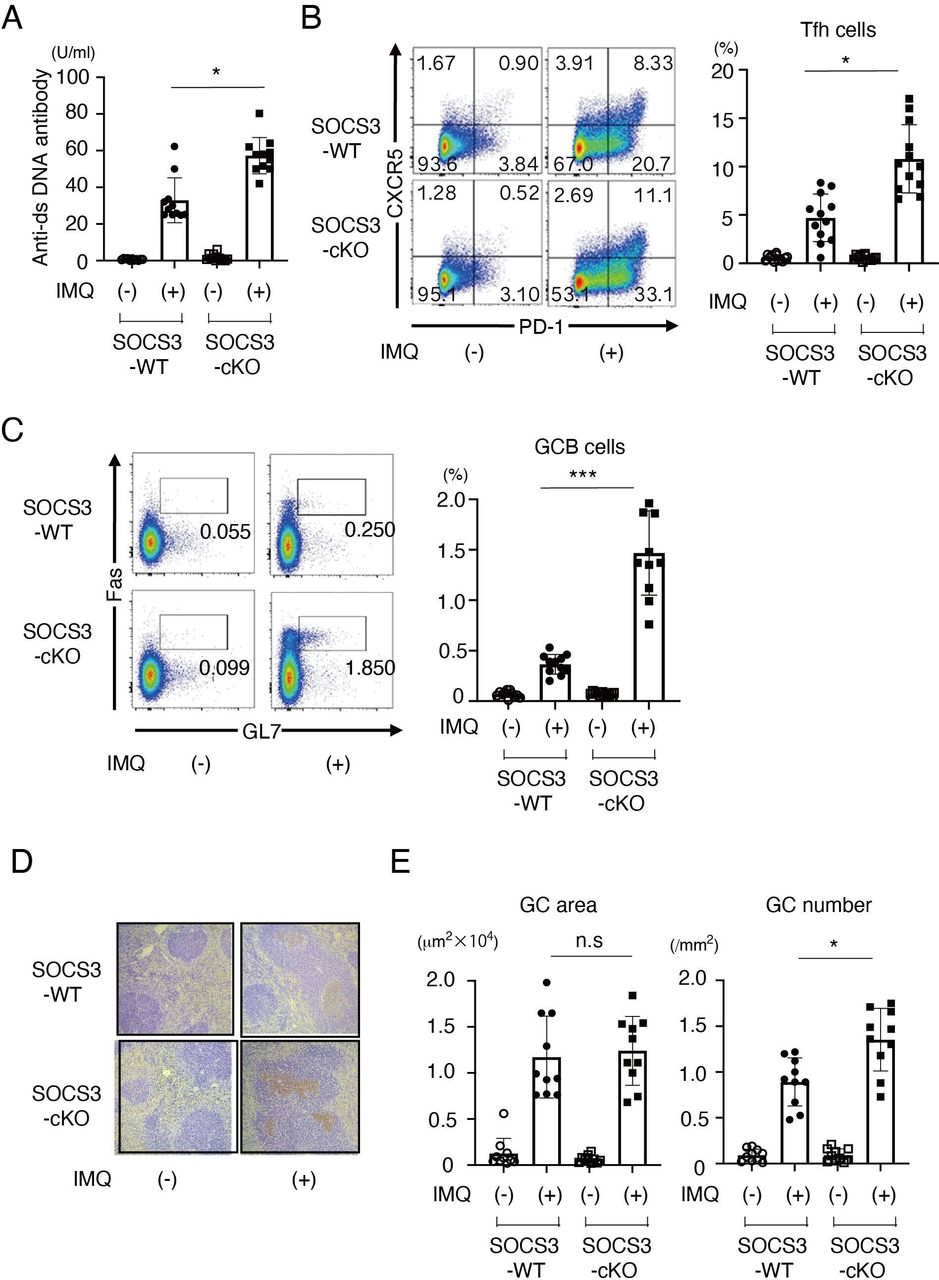
Since autoantibody production is associated with the pathogenesis of SLE, we next quantified the levels of anti-dsDNA antibody, a representative autoantibody in SLE, in SOCS3-cKO mice and SOCS3-WT mice in the imiquimod-induced lupus model. We found that serum levels of anti-dsDNA antibody were higher in SOCS3-cKO mice than those in SOCS3-WT mice (figure 3A) on imiquimod administration. As expected, in the absence of imiquimod administration, serum levels of anti-dsDNA antibody were very low in both SOCS3-cKO mice and SOCS3-WT mice (figure 3A).

Autoantibody production is increased in SOCS3-cKO mice. SOCS3-cKO mice and SOCS3-WT mice were treated with or without topical imiquimod as described in the Methods section. (A) Anti-dsDNA antibody levels in sera in SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. Data are means±SEM for 11 mice in each group. Data are compiled from two independent experiments. *p<0.05. (B) Shown are representative dot plots of CXCR5 vs PD-1 gated on CD4+ T cells (left) and frequency of Tfh cells (CXCR5+ PD-1+ cells) (right) in spleen in SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. Data are means±SEM for 11 mice in each group. Data are compiled from two independent experiments. *p<0.05. (C) Shown are representative dot plots of Fas vs GL7 gated on B220+ B cells (left) and frequency of GC B cells (Fas+ GL7+ cells) (right) in spleen in SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. Data are means±SEM for 11 mice in each group. Data are compiled from two independent experiments. ***p<0.001. (D) Representative photographs of spleen sections stained with PNA at 8 weeks. (E) Shown are means±SEM of average GC areas (square millimetres × 104) (left) and of the number of GC formation (per square millimetre) (right). Data are means±SEM for 10 mice in each group. Data are compiled from two independent experiments. *p<0.05. Anti-dsDNA, anti-double-strand DNA; GC, germinal centre; IMQ, imiquimod; KO, knockout; PD-1, programmed cell death-1; PNA, peanut agglutinin; SOCS, suppressor of cytokine signalling; WT, wild type.
Tfh cells play a central role in GC formation and high-affinity antibody production by inducing the differentiation of plasma cells and memory B cells.19 Recent studies have shown that the frequency of Tfh cells is increased in patients with SLE.20 21 We, therefore, investigated the frequencies of Tfh cells and GC B cells as well as GC formation in the spleen in SOCS3-cKO mice and SOCS3-WT mice in the imiquimod-induced lupus model. The frequencies of Tfh cells (figure 3B) and GC B cells (figure 3C), as well as GC formation in the spleen (figure 3D,E), were significantly increased in SOCS3-cKO mice compared with those in SOCS3-WT mice in the imiquimod-induced lupus model.
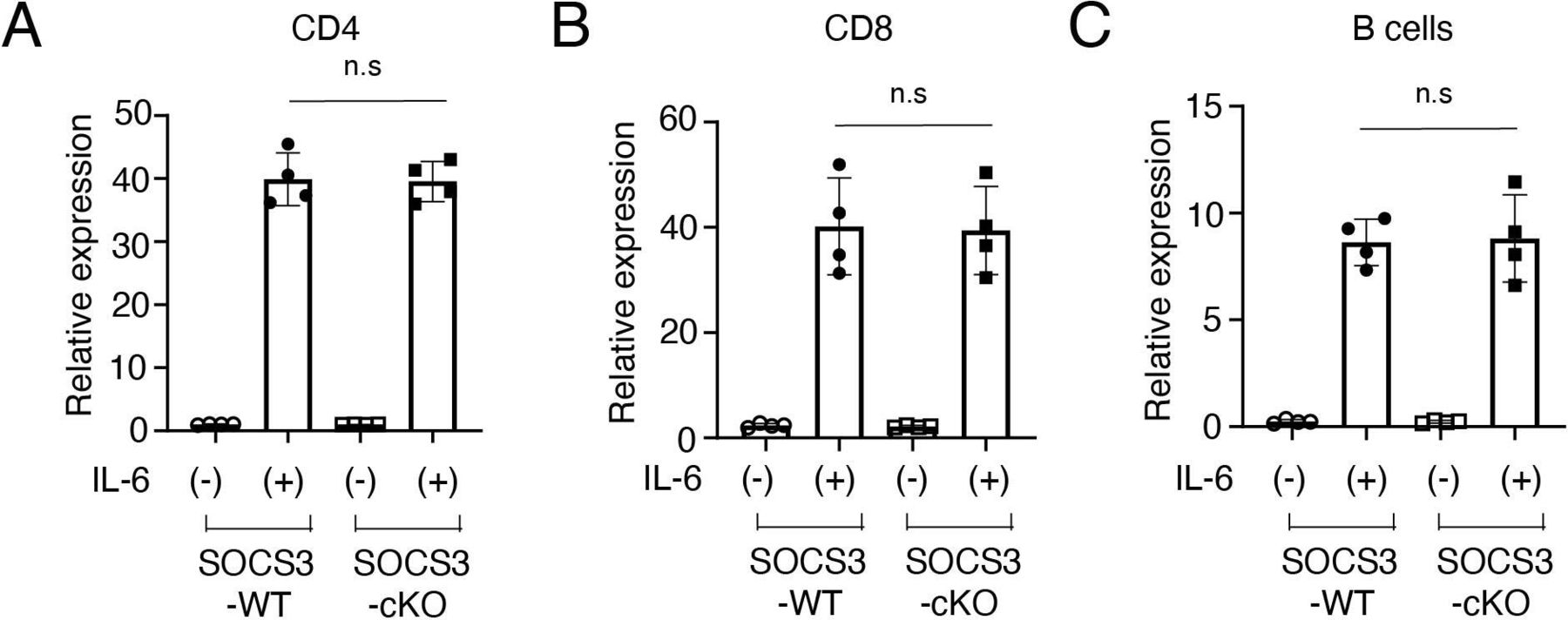
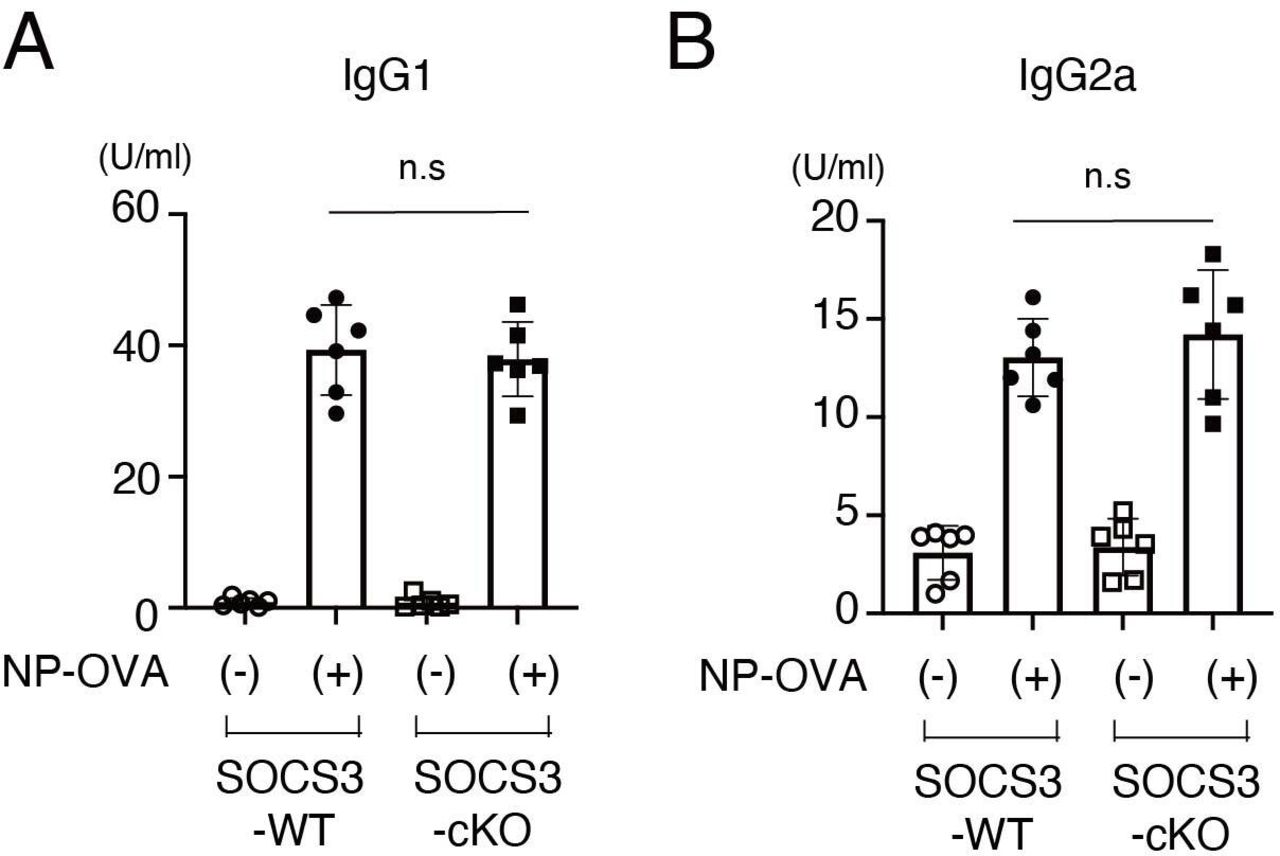
To exclude the possibility that Cre recombinase is expressed in T cells and B cells in podocin-Cre mice and SOCS3 expression is deleted in T cells and B cells in SOCS3-cKO mice, we examined mRNA expression of SOCS3 in IL-6-stimulated CD4+ T cells, CD8+ T cells and B cells in SOCS3-cKO mice and SOCS3-WT mice. SOCS3 expression was induced similarly between these mice in CD4+ T cells, CD8+ T cells and B cells on IL-6 stimulation (figure 4A–C). Moreover, we found that on immunisation with NP-OVA, the production of NP-specific IgG1 and IgG2a was induced similarly between SOCS3-cKO mice and SOCS3-WT mice (figure 5). These results suggest that the loss of SOCS3 expression in podocytes rather than immune cells causes the increase of Tfh cells and GC B cells and the enhanced anti-dsDNA antibody production in the imiquimod-induced lupus model.
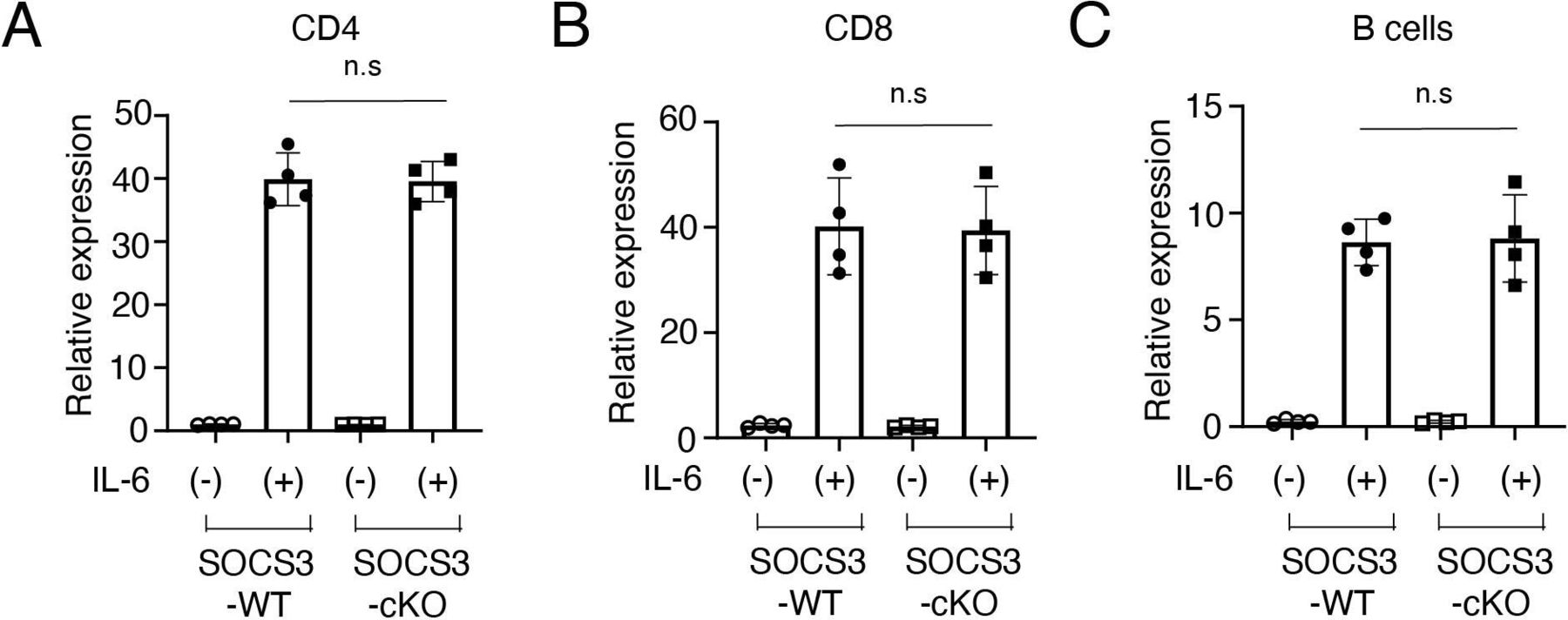
SOCS3 expression in CD4+ T cells, CD8+ T cells and B cells in SOCS3-cKO mice. Splenocytes were obtained from 8-week-old SOCS3-cKO mice and SOCS3-WT mice. (A) CD4+ T cells, (B) CD8+ T cells and (C) B cells were isolated as described in the Methods section,and were stimulated with or without IL-6 for 2 hours. The expression levels of SOCS3 were evaluated by qPCR analysis. Data are compiled of four independent experiments. IL-6, interleukin 6; KO, knockout; SOCS, suppressor of cytokine signalling; WT, wild type.

NP-OVA-induced antibody production in SOCS3-cKO mice. Eight-week-old SOCS3-cKO mice and SOCS3-WT mice were immunised intraperitoneally with NP-OVA as described in the Methods section. Seven days after the immunisation, serum levels of NP-specific IgG1 (A) and IgG2a (B) were quantified by ELISA. Data are means±SEM for six mice in each group. Data are compiled from two independent experiments. IgG, immunoglobulin G; KO, knockout; NP-OVA, 4-hydroxy-. 3- nitrophenylacetyl-ovalbumin; SOCS, suppressor of cytokine signalling; WT, wild type.
SOCS3 suppresses IL-6 expression in podocytes
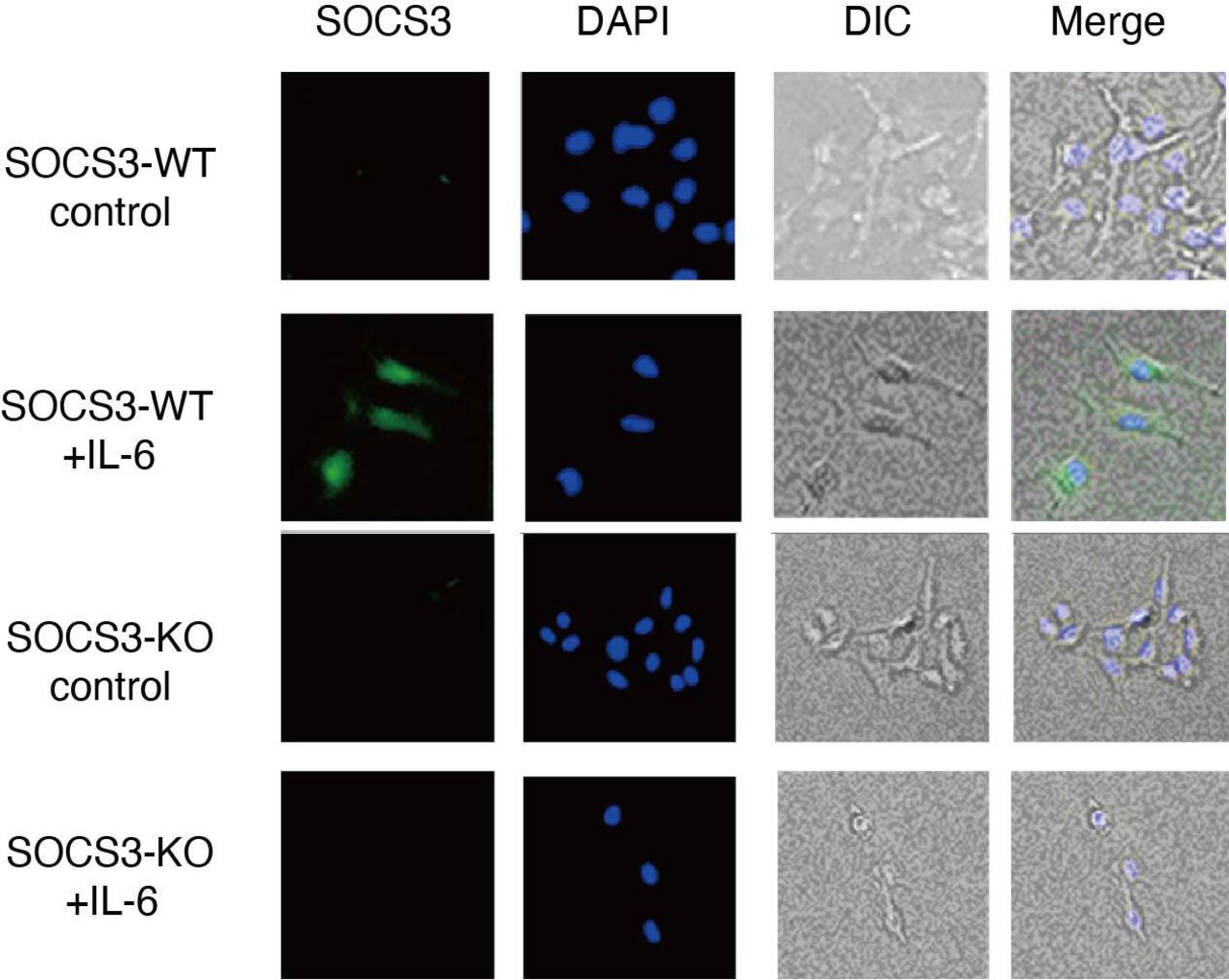
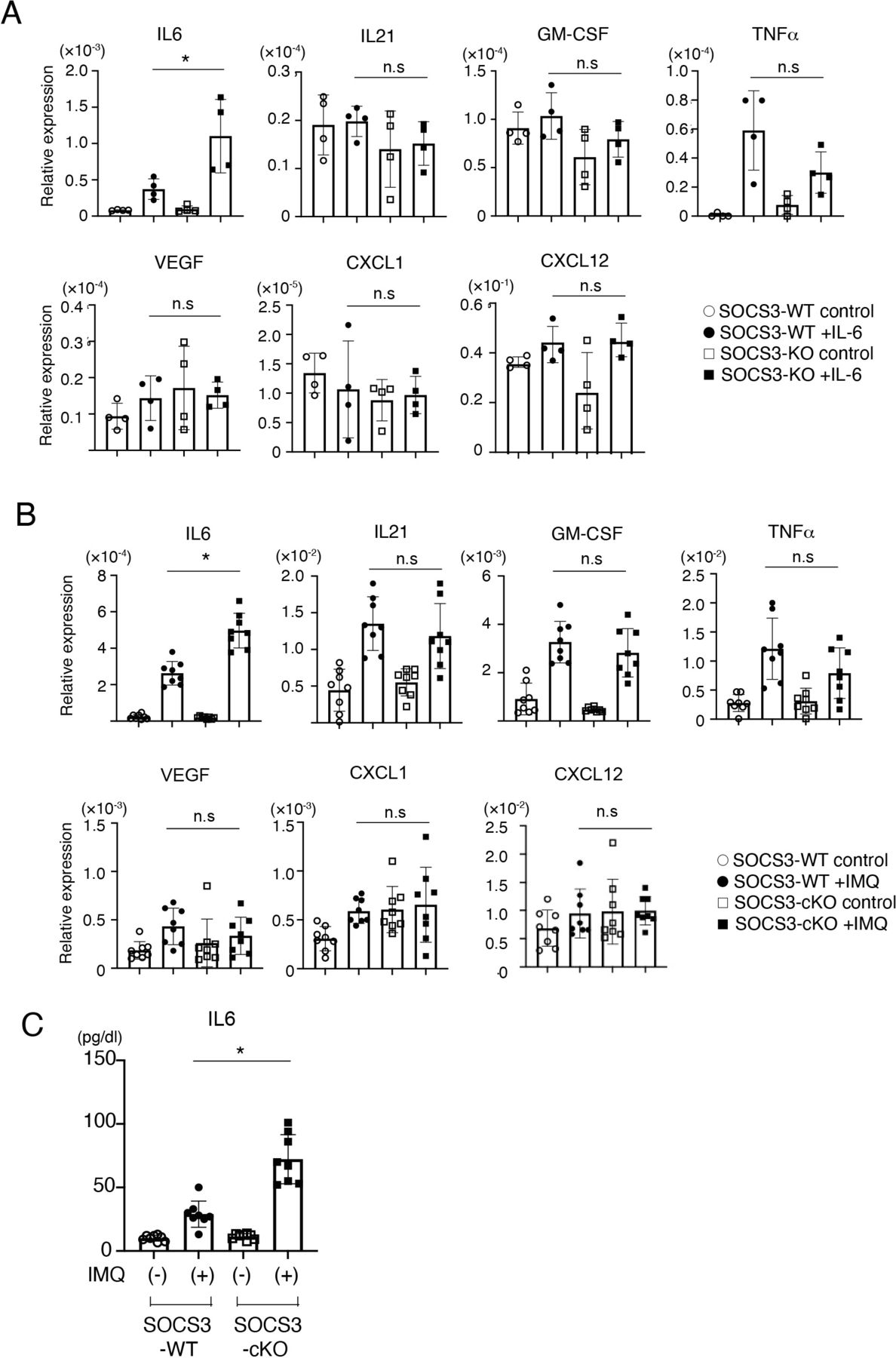
In addition to the barrier function, a recent study has shown that podocytes are involved in causing glomerulonephritis by producing proinflammatory cytokines/chemokines such as TNF-α, IL-6, IL-8, VEGF, GM-CSF and M-CSF.6 We, therefore, sought to analyse the expression of proinflammatory cytokines/chemokines in a SOCS3-deficient podocyte cell line in which SOCS3 expression is deleted by CRISPR-Cas9-mediated gene targeting (figure 6). Among proinflammatory cytokines/chemokines we examined, the expression of IL-6 was increased in the SOCS3-deficient podocyte cell lines compared with that in the control podocyte cell lines on IL-6 stimulation (figure 7A). In addition, we analysed the expression of these cytokines and chemokines in glomeruli isolated from SOCS3-cKO mice and SOCS3-WT mice in the imiquimod-induced lupus model and found that mRNA levels of IL-6 were higher in glomeruli in SOCS3-cKO mice than those in SOCS3-WT mice (figure 7B). We also quantified serum levels of IL-6 in the imiquimod-induced lupus model and found that the levels of IL-6 were higher in SOCS3-cKO mice than those in SOCS3-WT mice (figure 7C). These results suggest that SOCS3 deficiency in podocytes results in increased expression of IL-6 in the imiquimod-induced lupus model.
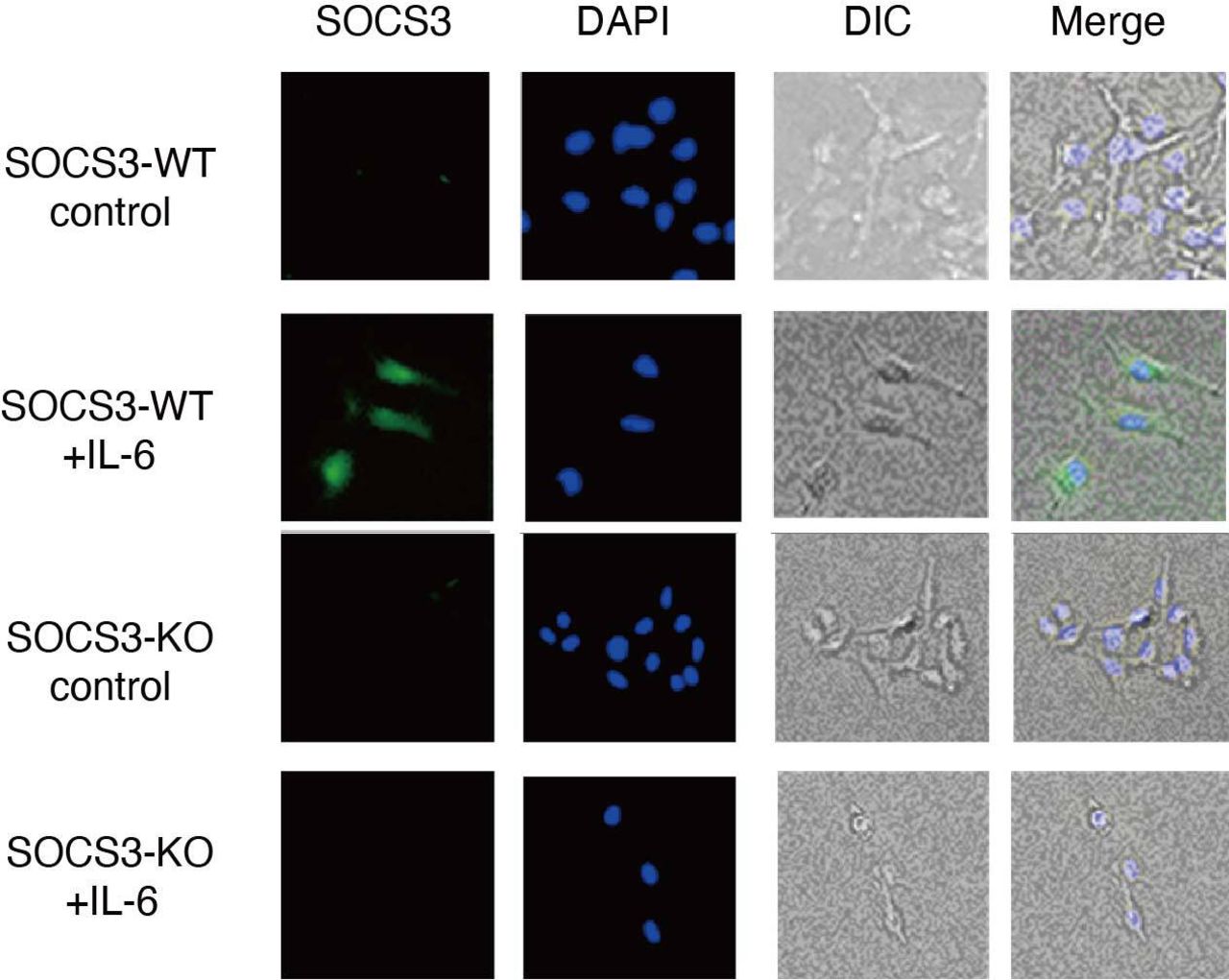
CRISPR-Cas9-mediated gene deletion of SOCS3 in a murine podocyte cell line was performed as described in the Methods section. Cells were stimulated with or without IL-6 for 2 hours and were stained with anti-SOCS3 antibody and DAPI. Shown are representative photographs. DAPI, 4',6-diamidino-2-phenylindole; DIC, differential interference contrast; IL-6, interleukin 6; KO, knockout; SOCS, suppressor of cytokine signalling; WT, wild type.

SOCS3 expressed in podocytes suppresses local and systemic IL-6 expression. (A) CRISPR-Cas9-mediated gene deletion of SOCS3 in a murine podocyte cell line was performed as described in the Methods section. Cells were stimulated with or without IL-6 for 2 hours, and the expression levels of indicated genes were evaluated by qPCR analysis. Data are compiled of four independent experiments. *p<0.05. (B) SOCS3-cKO mice and SOCS3-WT mice were treated with or without topical imiquimod for 8 weeks as described in the Methods section. Glomeruli were isolated and the expression levels of indicated genes in the glomeruli were evaluated by qPCR analysis. Data are means±SEM for eight mice in each group. Data are compiled from two independent experiments. *p<0.05. (C) Serum levels of IL-6 in SOCS3-cKO mice and SOCS3-WT mice at 8 weeks. Data are means±SEM for eight mice in each group. Data are compiled from two independent experiments. *p<0.05. CXCL, chemokine (C-X-C motif) ligand; GM-CSF, granulocyte macrophage colony-stimulating factor; IL6, interleukin 6; IMQ, imiquimod; KO, knockout; SOCS, suppressor of cytokine signalling; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor; WT, wild type.
SOCS3 and IL-6 are expressed in podocytes in MRL/lpr mice
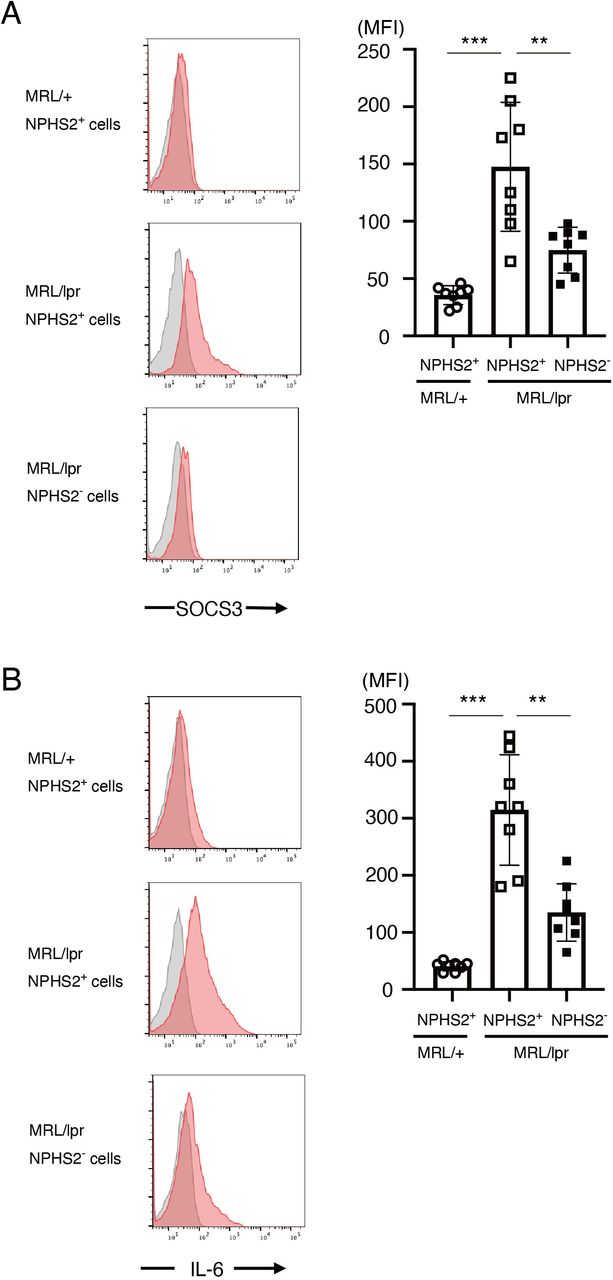
To clarify whether our findings are unique to the imiquimod-induced lupus model or not, we analysed SOCS3 and IL-6 expression in podocytes in a spontaneous murine lupus model, MRL/lpr mice. We found that SOCS3 expression in NPHS2(podocin)+ cells was higher in 6-month-old MRL/lpr mice than in control MRL/+mice (figure 8A). We also found that SOCS3 expression was higher in NPHS2+ cells than in NPHS2− cells in MRL/lpr mice (figure 8A). Analogously, IL-6 expression was higher in NPHS2+ cells in MRL/lpr mice than in NPHS2− cells in MRL/lpr mice or NPHS2+ cells in MRL/+mice (figure 8B). These results suggest that SOCS3 and IL-6 are expressed in podocytes not only in the imiquimod-induced lupus model but also in the spontaneous murine lupus model MRL/lpr mice.

SOCS3 and IL-6 are expressed in podocytes in MRL/lpr mice. Single-cell suspensions were prepared from isolated glomeruli in 6-month-old MRL/lpr mice and control MRL/+mice. Intracellular staining of SOCS3 and IL-6, together with staining of podocin (NPHS2), was performed as described in the Methods section. (A) Left panels are the representative SOCS3 staining (red) and isotype-matched control staining (grey) gated on NPHS2+ cells in control MRL/+mice and MRL/lpr mice and on NPHS2− cells in MRL/lpr mice. Data are means±SEM of mean fluorescence intensity (MFI) of SOCS3 for eight mice in each group. Data are compiled from two independent experiments. ***p<0.001, **p<0.01. (B) Left panels are the representative IL-6 staining (red) and isotype-matched control staining (grey) gated on NPHS2+ cells in control MRL/+mice and MRL/lpr mice and on NPHS2− cells in MRL/lpr mice. Data are means±SEM of MFI of IL-6 for eight mice in each group. Data are compiled from two independent experiments. ***p<0.001, **p<0.01. IL-6, interleukin 6; SOCS,suppressor of cytokine signalling.
SOCS3 deficiency in podocytes results in reduced survival in the imiquimod-induced lupus model
We finally compared the survival rate of SOCS3-WT mice and SOCS3-cKO mice in the imiquimod-induced lupus model. As shown in figure 9, SOCS3-cKO mice exhibited a reduced survival rate compared with SOCS3-WT mice. Without the imiquimod administration, no deaths occurred even in SOCS3-cKO mice in this experimental period (data not shown). These results suggest that SOCS3 expressed in podocytes protects mice from death in the imiquimod-induced lupus model.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SOCS3-cKO mice exhibit reduced survival rates in the imiquimod-induced lupus model. SOCS3-cKO mice and SOCS3-WT mice were treated with topical imiquimod as described in the Methods section. Shown are cumulative survival rates of SOCS3-cKO mice and SOCS3-WT mice following topical administration of imiquimod on the ear skin three times per week, n=12, each. KO, knockout; SOCS, suppressor of cytokine signalling; WT, wild type.
Discussion
In the present study, we show that SOCS3 is preferentially expressed in glomerular podocytes in the imiquimod-induced lupus model and MRL/lpr mice and is induced in podocyte cell lines on IL-6 stimulation. In the imiquimod-induced lupus model, SOCS3-cKO mice, which lack the expression of SOCS3 in podocytes but not in immune cells, exhibited severe glomerulonephritis, high levels of serum creatinine and urine albumin and decreased survival rates compared with control SOCS3-WT mice. Unexpectedly, serum levels of anti-dsDNA antibodies, GC formation and the numbers of Tfh cells and GC B cells were elevated in SOCS3-cKO mice compared with those in SOCS3-WT mice. Importantly, serum levels of IL-6, as well as the expression levels of IL-6 mRNA in glomeruli, were elevated in SOCS3-cKO mice, and IL-6-induced IL-6 expression was increased in SOCS3-deficient podocyte cell lines. These results suggest that SOCS3 expressed in podocytes plays protective roles for the development of glomerulonephritis and inhibits autoantibody production in the imiquimod-induced lupus model, presumably by suppressing a vicious cycle of IL-6 production in podocytes.
SLE is a prototypic systemic autoimmune disease mediated by dysregulated T cells and B cells. The dysregulated immune responses lead to the production of various autoantibodies including anti-dsDNA antibody and the generated autoantibody–autoantigen complexes damage multiple organs including kidneys.22–24 Recent studies have suggested that Tfh cells, a subset of CD4+ T cells essential for the GC formation and high-affinity antibody production via the induction of plasma cell and memory B cell differentiation, play pathogenetic roles in SLE by activating autoantibody-producing B cells.19 25 26 Moreover, it has been reported that the number of Tfh cells is increased in peripheral blood in patients with SLE.27 These studies suggest that Tfh cells play crucial roles in the pathogenesis of SLE by inducing autoantibody production.
We show here that SOCS3 deficiency in podocytes results in severe glomerulonephritis and decreased survival rates in the imiquimod-induced lupus model. We also show that Tfh cells (figure 3B) and GC B cells (figure 3C) were increased in SOCS3-cKO mice compared with those in control SOCS3-WT mice in the imiquimod-induced lupus model. In addition, serum levels of anti-dsDNA antibody were higher in SOCS3-cKO mice than those in SOCS3-WT mice in the imiquimod-induced lupus model (figure 3A). Taken together, our data suggest that loss of SOCS3 expression in podocytes not only aggravates glomerulonephritis but also increases the frequencies of Tfh cells and GC B cells, resulting in hyperproduction of autoantibodies in the imiquimod-induced lupus model.
The mechanisms underlying the increased Tfh cells and GC B cells and the enhanced autoantibody production in SOCS3-cKO mice remain unclear. Because SOCS3 was normally induced in T cells and B cells in SOCS3-cKO mice (figure 4), it is unlikely that leaked expression of Cre recombinase in podocin-Cre mice caused the deletion of SOCS3 gene in T cells and B cells in SOCS3-cKO mice. We also found that systemic immune responses on immunisation with NP-OVA were normal in SOCS3-cKO mice (figure 5). These findings support the notion that T cell and B cell responses are not primarily responsible for the increased Tfh cells and GC B cells and the enhanced autoantibody production in SOCS3-cKO mice.
It is well-known that IL-6 promotes Tfh cell differentiation by inducing the expression of BCL6, a master transcription factor of Tfh cells.28 IL-6 is a pleiotropic cytokine, which influences the growth and differentiation of various cell types, induces antibody production and mediates inflammatory responses. Consequently, IL-6 is deeply involved in the pathogenesis of many inflammatory and autoimmune diseases.29 Indeed, it has been shown that serum IL-6 levels are elevated in patients with SLE and that IL-6 levels correlate with disease activity.30 Moreover, it has been shown that polymorphisms in the IL-6 gene were associated with SLE susceptibility.31 Furthermore, Shirota et al32 have shown that in vivo blockade of the IL-6 signalling by tocilizumab, a humanised monoclonal antibody against the IL-6 receptor, decreases the frequencies of activated CD4+ T cells, activated B cells and plasmablasts/plasma cells in patients with SLE. Although it remains unclear whether in vivo blockade of the IL-6 signalling affects the differentiation of Tfh cells and GC B cells in patients with SLE, a recent murine study has shown that the blockade of IL-6 signalling results in the reduction of Tfh cells in lupus-prone BXSB.Yaa mice.33 These findings suggest that IL-6 plays a pivotal role in the expansion of Tfh cells in SLE, resulting in the increase in GC B cells and plasma cells and the production of autoantibody. In this regard, we found here that serum levels of IL-6 were elevated in SOCS3-cKO mice compared with those in control SOCS3-WT mice in the imiquimod-induced lupus model (figure 7C). It has been reported that a positive feedback loop of IL-6 signalling between non-immune cells and immune cells leads to excessive IL-6 expression.34 Considering these findings, the deficiency of SOCS3 in podocytes may create the positive feedback loop of IL-6 production, leading to the increase in Tfh cells and GC B cells and subsequent upregulation of anti-dsDNA antibody production in this lupus model.
Recently, several studies have examined whether IL-6 signalling could be a therapeutic target in patients with SLE. First, the effect of tocilizumab was examined in an open-label phase I dosage-escalation study.35 Although tocilizumab improved disease activity and decreased the levels of anti-dsDNA antibodies in patients with SLE, the patients treated with tocilizumab suffered from infections frequently.35 On the contrary, sirukumab, a fully human anti-IL-6 monoclonal antibody, did not demonstrate the anticipated efficacy in patients with SLE with active LN.36 These studies suggest that although IL-6 signalling could be a therapeutic target in SLE, new approaches are required. Our data suggest that the induction of SOCS3 in podocytes could be a therapeutic approach for treating SLE via the blockade of IL-6 signalling.
Data availability statement
Data are available in a public, open access repository. Kotaro Suzukisuzuki_k@faculty.chiba-u.jp.
Ethics statements
Ethics approval
The animal experiments in this study were approved by the Animal Committee of Graduate School of Medicine, Chiba University (approval ID: A1-69).
Acknowledgments
We thank Ms M Yoshino and Ms K Nemoto (Chiba University) for animal care and technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KoS conceived and designed the experiment. MF, KaS, SK, KI, HY, SM, AI, ST, TI, AS, DN, HW, YuM, YoM and MT were involved in the experiments. KoS, KA and HN were involved in the data interpretation and writing of the manuscript. The authors read and approved the final manuscript.
Funding This research was supported in part by grants-in aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government and by LGS (Leading Graduate School) Programme, MEXT, Japan.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.