Article Text

Abstract
Objective To assess the appropriateness of expanded serological activity eligibility criteria for belimumab use in the UK systemic lupus erythematosus (SLE) population (and possibly other countries), which includes patients with either anti-double-stranded DNA (anti-dsDNA) positivity or hypocomplementaemia rather than both criteria.
Methods This post-hoc analysis used data from three randomised, double-blind, placebo-controlled phase III belimumab trials: BLISS-52 (BEL110752; NCT00424476), BLISS-76 (BEL110751; NCT00410384) and BLISS-SC (BEL112341; NCT01484496). Patients with SLE were stratified by high disease activity (HDA): HDA1, Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI) score ≥10, low complement and positive anti-dsDNA; and HDA2, SELENA-SLEDAI ≥10 and either low complement or positive anti-dsDNA.
Results This analysis included 660 HDA1 patients (n=396 on intravenous treatment; n=264 on subcutaneous treatment) and 969 HDA2 patients (n=532 on intravenous treatment; n=437 on subcutaneous treatment). Significant improvements were observed at week 52 with belimumab versus placebo, irrespective of subgroups or drug formulations, in SLE Responder Index (SRI) 4 response (OR (95% CI): HDA1 intravenous 2.7 (1.8 to 4.1); HDA2 intravenous 2.3 (1.61 to 3.26); HDA1 subcutaneous 2.2 (1.22 to 3.85); HDA2 subcutaneous 1.8 (1.17 to 2.74)); proportion of patients achieving ≥4-point reduction in SELENA-SLEDAI score (OR (95% CI): HDA1 intravenous 2.6 (1.7 to 3.9); HDA2 intravenous 2.1 (1.49 to 3.03); HDA1 subcutaneous 2.3 (1.30 to 4.14); HDA2 subcutaneous 1.9 (1.21 to 2.84)); patients with no worsening in Physician Global Assessment (OR (95% CI): HDA1 intravenous 2.0 (1.3 to 3.1); HDA2 intravenous 1.7 (1.17 to 2.45); HDA1 subcutaneous 2.3 (1.18 to 4.40); HDA2 subcutaneous 1.8 (1.11 to 2.92)); and risk of severe flares (HR (95% CI): HDA1 intravenous 0.6 (0.37 to 0.81); HDA2 intravenous 0.6 (0.43 to 0.86); HDA1 subcutaneous 0.52 (0.30 to 0.92); HDA2 subcutaneous 0.59 (0.37 to 0.94)).
Conclusion Broadening the HDA population to include either low complement or positive anti-dsDNA, rather than both, would enable more UK patients to receive SLE treatment and experience improved clinical outcomes.
- lupus erythematosus
- systemic
- quality of lIfe
- therapeutics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Clinical trials have demonstrated a favourable benefit–risk profile with belimumab for treatment of SLE; post-hoc analyses have also indicated that patients with high disease activity (HDA) may receive particular benefit from belimumab.
UK treatment guidelines require patients to have evidence of serological activity (low complement and positive anti-double-stranded DNA (anti-dsDNA)) and a score ≥10 in the Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI) instrument (or equivalent) to be eligible for belimumab treatment through the National Health Service.
What does this study add?
In this post-hoc analysis, belimumab showed increased efficacy versus placebo across 1125 patients with SLE and HDA pooled from the BLISS-52, BLISS-76 and BLISS-SC studies.
Belimumab demonstrated similar clinically meaningful efficacy when requiring patients to have at least one biomarker (either low complement or positive anti-dsDNA) along with a SELENA-SLEDAI score ≥10 as baseline predictors of response, compared with the current criteria of requiring patients to always have both biomarkers as well as SELENA-SLEDAI score ≥10.
How might this impact on clinical practice or future developments?
These data suggest that expanding the UK criteria to include patients with SLE with HDA with low complement or positive anti-dsDNA, as opposed to the current requirement for both, would allow more patients to receive belimumab treatment and thereby improve clinical outcomes.
Introduction
In two pivotal phase III, randomised, double-blind, placebo-controlled studies (BLISS-52 and BLISS-76), belimumab (intravenous 10 mg/kg monthly) plus standard therapy for systemic lupus erythematosus (SLE) demonstrated a favourable benefit–risk profile for treatment of SLE.1 2 Belimumab-treated patients also showed an improvement in health-related quality of life measures, which were consistent with the observed reduction in disease activity.3 A post-hoc analysis of these two studies indicated that a pooled subgroup of patients with high disease activity (HDA), denoted by the Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI) score of ≥10, anti-double-stranded DNA (anti-dsDNA) positivity, hypocomplementaemia or requiring corticosteroid treatment, may derive particular benefit from treatment with belimumab in addition to standard therapy, compared with standard therapy alone.4 In addition, an open-label extension study (GSK Study 112233; NCT00724867) of intravenous belimumab in patients who completed BLISS-76 also showed significant improvements in disease activity as well as suggested a corticosteroid-sparing effect.5
A phase III study of a subcutaneous formulation of belimumab (200 mg weekly) plus standard therapy (BLISS-SC) showed results consistent with intravenous treatment, with the enhanced treatment option of subcutaneous administration.6 Additionally, a 6-month, open-label extension of this study showed a favourable safety profile and maintained efficacy.7 A post-hoc analysis of patients with HDA from the BLISS-SC study has also been conducted. Patients with hypocomplementaemia and anti-dsDNA-positive SLE showed reduced disease activity and improved levels of fatigue in those receiving belimumab versus placebo.8
In the European Union, belimumab is indicated as an add-on therapy in adults with active, autoantibody-positive SLE with a high degree of disease activity (eg, low complement and positive anti-dsDNA) despite standard therapy.9 In the UK, the National Institute for Health and Care Excellence guidelines stipulate that, in addition to evidence of serological activity (low complement and positive anti-dsDNA), patients with SLE must have a score ≥10 in the SELENA-SLEDAI, or equivalent SLEDAI-2000 instrument, to be eligible for treatment with belimumab covered by the National Health Service in England and Wales.10 As patients must satisfy three criteria to become eligible for treatment with belimumab in the UK, it may narrow the population that could benefit from treatment.11 This post-hoc analysis of the pooled BLISS-52, BLISS-76 studies and the BLISS-SC study investigates the appropriateness of expanded serological activity eligibility criteria for belimumab use in the UK population, which includes patients with either low complement or anti-dsDNA positivity, rather than requiring both criteria.
Methods
Study design and patient population
This was a post-hoc analysis (GSK Study 207878) of data from three randomised, double-blind, placebo-controlled phase III trials: BLISS-52 (BEL110752; NCT00424476), BLISS-76 (BEL110751; NCT00410384) and BLISS-SC (BEL112341; NCT01484496). The BLISS trials were conducted according to the principles of the Declaration of Helsinki and appropriate ethical approvals were obtained.1 2 6
BLISS-52 (52 weeks) and BLISS-76 (76 weeks) were parallel-group studies investigating the efficacy and safety of belimumab administered by intravenous infusion in adult patients with active SLE.1 2 The study designs and methods have been described previously, and the primary endpoint for both studies was response rate at week 52, assessed by SLE Responder Index 4 (SRI4).1 2 Patients were randomly assigned to one of three treatment groups: belimumab 1 mg/kg, belimumab 10 mg/kg or placebo. Each treatment was administered in addition to standard therapy. Belimumab or placebo was administered over a 1-hour period on days 0, 14 and 28, and then every 28 days. Standard therapy dose could be adjusted only up to week 24. Changes in the total dose of systemic corticosteroids were permitted until week 24; however, the total systemic dose must have returned to within 25% or 5 mg over the baseline dose, whichever was higher, by that visit. After week 24, the dose had to be within 25% or 5 mg of the baseline dose, whichever was higher. No increase in steroids was allowed in the 8 weeks preceding the week 52 (weeks 44–52) and week 76 (weeks 68–76; BLISS-76 only) visits. New antimalarial agents were permitted up to week 16. No new immunosuppressive or immunomodulatory agents were permitted after day 0 for either study; dose increase of existing agents was only permitted between baseline (day 0) and week 16 visit.
Patients ≥18 years of age were required to have a diagnosis of SLE classified according to the American College of Rheumatology criteria,12 13 with positive ANA (titre ≥1:80) and/or anti-dsDNA antibodies (≥30 IU/mL) and a SELENA-SLEDAI score of ≥6 at screening. Patients also had a stable treatment regimen with fixed doses of prednisone (0–40 mg/day) or non-steroidal anti-inflammatory, antimalarial or immunosuppressive drugs for at least 30 days before the first study dose. Severe active lupus nephritis or central nervous system (CNS) lupus and pregnancy were exclusion criteria. Patients who had previously been treated with any B lymphocyte-targeted drug (including rituximab) at any time, intravenous cyclophosphamide within 6 months of enrolment, and intravenous immunoglobulin or prednisone (>100 mg/day) within 3 months were also excluded.
BLISS-SC was a 52-week study designed to evaluate the efficacy and safety of subcutaneous belimumab in adult patients with active SLE.6 Briefly, in addition to stable standard therapy, after a screening period (up to 35 days), eligible patients were randomised 2:1 to receive belimumab 200 mg subcutaneously or placebo, weekly, in the 52-week, double-blind treatment period. Patients ≥18 years of age with a diagnosis of SLE with ANA and/or anti-dsDNA antibodies and a score of ≥8 in SELENA-SLEDAI at screening were eligible. Patients were excluded if they had severe lupus kidney disease (proteinuria >6 g/24 hours or equivalent according to a spot urinary protein to creatinine ratio or a serum creatinine level >2.5 mg/dL) or severe CNS lupus.
Post-hoc analysis
The current post-hoc analysis investigated two HDA patient subgroups with SELENA-SLEDAI score ≥10 at baseline: HDA1, with low complement (C3 or C4) and positive anti-dsDNA; and HDA2, with low complement and/or positive anti-dsDNA. Post-hoc analyses of the primary endpoint for both subgroups were carried out separately for the intravenous and subcutaneous data sets. For the intravenous data set, only patients who received placebo and belimumab 10 mg/kg intravenously were included in these analyses.
Endpoints and assessments
The primary endpoint of this current analysis was SRI4 response rate at week 52, a composite index consistent with the primary efficacy endpoint for the belimumab phase III clinical trials. An interim analysis of the BLISS-76 study was conducted at week 52. SRI4 responders had ≥4-point reduction in SELENA-SLEDAI score, no new British Isles Lupus Assessment Group (BILAG) A organ domain scores and <2 new BILAG B organ domain scores, and no worsening (increase <0.3 from baseline) in Physician Global Assessment (PGA) score at week 52 compared with baseline. Endpoints that supported the primary endpoint included the SRI4 by visit and SRI4 components by visit.
Secondary endpoints included the SRI4 response rate at week 76 (patients from BLISS-76 only) and the Short Form 36-Item (SF-36) Health Survey questionnaire, Physical Component Summary (PCS) score at week 52 (intravenous studies only), and reduction in prednisone dose (proportion of patients among those receiving >7.5 mg/day at baseline who experienced a mean corticosteroid dose reduction of ≥25% mg/day at baseline to ≤7.5 mg/day during weeks 40–52). Time to first SLE flare (any) and time to first severe SLE flare as measured by SELENA-SLEDAI Flare Index (SFI; weeks 0–52; as defined in Petri et al14) were also assessed, with hazard ratio (HR) used to indicate flare risk.
Statistical analysis
The primary endpoint and supporting results for the intravenous data were analysed using a logistic regression model adjusted for randomisation stratification factor of race (black African ancestry or indigenous-American descent vs other). The primary endpoint and supporting results for the subcutaneous data were also analysed using a logistic regression model adjusted for randomisation stratification factor of race (black African ancestry or indigenous-American descent vs other), as all other stratification factors (baseline SELENA-SLEDAI score ≤9 vs ≥10) and baseline complement levels (low C3 and/or C4 vs no low C3 or C4) were not appropriate given the criteria for the subcutaneous subgroups considered here. Additional covariates used in the analyses were baseline proteinuria levels (<2 vs ≥2 g/24-hour equivalent) for intravenous and subcutaneous subgroups, study (BLISS-76 vs BLISS-52) for intravenous subgroups only, baseline SELENA-SLEDAI score for subcutaneous subgroups only, and baseline values for the BILAG and PGA components of the primary endpoint. For subcutaneous data analysis, baseline SELENA-SLEDAI score was also included as a covariate. Secondary endpoints were analysed using a logistic regression model, analysis of covariance (ANCOVA) or a Cox proportional hazards model, as appropriate. For logistic regression analyses odds ratios (ORs) and 95% CI are presented. For ANCOVA, least squares means and standard errors (SEs) are presented, and for Cox proportional hazards model analyses HR and 95% CI are presented. Covariates were as specified above for the primary endpoint. Statistical testing was two-sided, with significance interpreted at p≤0.05. There were no multiplicity adjustments and nominal p values are presented.
Results
Patient population
The BLISS-52 and BLISS-76 studies included a total of 1125 patients (intravenous placebo, n=562; intravenous belimumab 10 mg/kg, n=563).1 2 The patient populations making up each of the subgroups are shown in online supplemental figure 1. Of these, 396 (35.2%; intravenous placebo, n=203; intravenous belimumab, n=193) were included in HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and 532 (47.3%; intravenous placebo, n=270; intravenous belimumab, n=262) were included in HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA). The BLISS-SC study included a total of 836 patients (subcutaneous placebo, n=280; subcutaneous belimumab 200 mg, n=556),6 of whom 264 (37.7%; subcutaneous placebo, n=78; subcutaneous belimumab, n=186) were included in HDA1 and 437 (62.3%; subcutaneous placebo, n=141; subcutaneous belimumab, n=296) were included in HDA2.
Supplemental material
Patient demographics and disease characteristics were similar between placebo and belimumab treatments for the intravenous and subcutaneous populations, as well as in HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) (table 1). In HDA2 there was an overlap of patients with positive anti-dsDNA (intravenous placebo=92.2%; intravenous belimumab=90.1%; subcutaneous placebo=90.8%; subcutaneous belimumab=94.6%) and low complement (intravenous placebo=83.0%; intravenous belimumab=83.6%; subcutaneous placebo=64.5%; subcutaneous belimumab=68.2%). Mean SELENA-SLEDAI scores at baseline were ≥12, fulfilling the inclusion criterion for HDA (SELENA-SLEDAI score ≥10), and were similar for all populations.
Patient demographics and characteristics at baseline
SRI4 response
Significant improvement in SRI4 was observed at week 52 with belimumab versus placebo in both subgroups (HDA1: SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA; and HDA2: SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) with both the intravenous and subcutaneous formulations (table 2). At week 52, more belimumab-treated patients, regardless of subgroups and formulations, achieved SRI4 response compared with placebo (HDA1 intravenous: 121 of 193 (62.7%) vs 77 of 203 (37.9%); HDA2 intravenous: 166 of 262 (63.4%) vs 117 of 270 (43.3%); HDA1 subcutaneous: 134 of 186 (72.0%) vs 44 of 78 (56.4%); HDA2 subcutaneous: 206 of 296 (69.6%) vs 79 of 141 (56.0%)) (table 2). For the intravenous formulation HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) subgroup, a statistically significant improvement in SRI4 response with belimumab versus placebo was observed at week 8 and was maintained between week 24 and week 52 (figure 1A). Similarly, for the intravenous formulation HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) subgroup, a statistically significant improvement in SRI4 response with belimumab versus placebo was maintained between week 16 and week 52, except for week 20 (figure 1B).

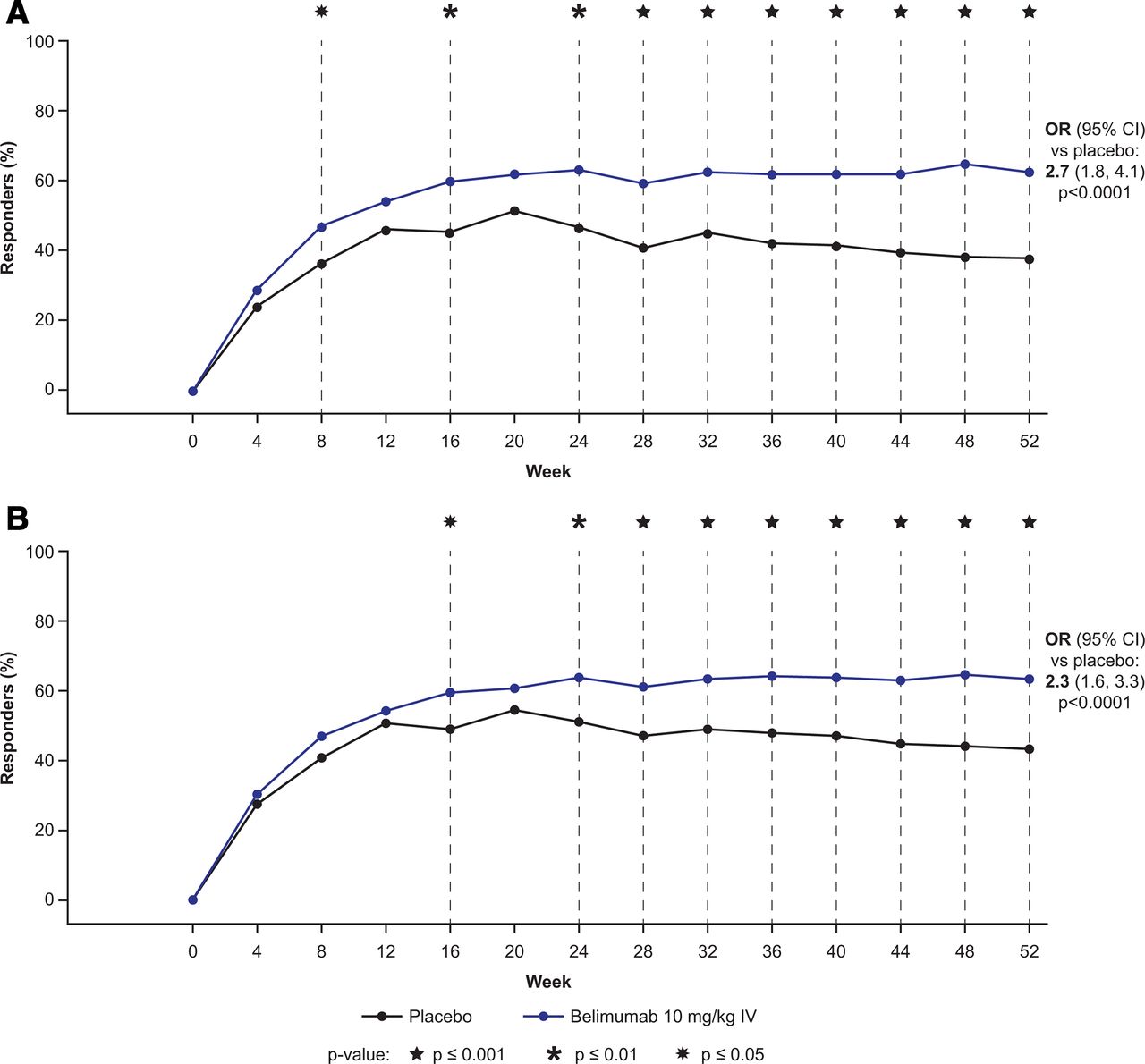{kind=link}
SRI4 response by visit through week 52 for (A) HDA1 intravenous subgroup and (B) HDA2 intravenous subgroup. HDA1: SELENA-SLEDAI score ≥10 at baseline with low complement and positive anti-dsDNA; HDA2: SELENA-SLEDAI score ≥10 at baseline with low complement and/or positive anti-dsDNA. dsDNA, double-stranded DNA; HDA, high disease activity; SELENA-SLEDAI, Safety of Estrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index; SRI4, SLE Responder Index 4.
SRI4 response rate and components of the SRI4 at week 52
Post-hoc analysis of the individual components of the primary endpoint also demonstrated significant improvements in belimumab-treated patients in SELENA-SLEDAI, BILAG and PGA parameters. At week 52, the proportion of patients achieving ≥4-point reduction in SELENA-SLEDAI score was significantly higher with belimumab versus placebo in both HDA subgroups for both intravenous and subcutaneous formulations (table 2). At week 52, a significantly greater proportion of patients in the HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) treatment groups (intravenous and subcutaneous) and the HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) intravenous subgroup reported no new 1A/2B BILAG domain scores compared with placebo (table 2). Although BILAG domain scores for the HDA2 group receiving the subcutaneous formulation did not significantly differ from placebo (p=0.0940), the OR (1.5 (95% CI 0.93 to 2.48)) was comparable with those of the HDA1 subcutaneous subgroup (2.0 (95% CI 1.02 to 3.78)) and the HDA1 and HDA2 intravenous groups (1.9 (95% CI 1.2 to 3.0) and 1.6 (95% CI 1.09 to 2.31), respectively) (table 2). A significantly higher proportion of patients in the HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) belimumab groups (both formulations) had no worsening in PGA versus placebo (table 2).
Secondary endpoints
The secondary endpoint, SRI4 response rate at week 76 (assessed for the BLISS-76 study only), was significantly greater with intravenous belimumab than with placebo for HDA1 intravenous (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA; 49.4% vs 31.3%, respectively; OR (95% CI) 2.1 (1.1 to 3.9), p=0.0188), but not for HDA2 intravenous (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA; 47.0% vs 37.0%, respectively; OR (95% CI) 1.5 (0.9 to 2.5), p=0.1288; table 3). There was a significant difference in the proportion of patients receiving intravenous belimumab versus placebo in their ability to reduce prednisone use by ≥25% from baseline to ≤7.5 mg/day during weeks 40–52 (HDA1 intravenous: 15.9% vs 7.1%, respectively; OR (95% CI) 2.4 (1.05 to 5.65), p=0.0389; HDA2 intravenous: 16.7% vs 6.7%, respectively; OR (95% CI) 2.6 (1.26 to 5.54), p=0.0104). The intravenous belimumab group also saw a reduction in time to any flares (HDA1 intravenous: HR (95% CI) 0.7 (0.56 to 0.88), p=0.0017; HDA2 intravenous: HR (95% CI) 0.8 (0.65 to 0.95), p=0.0131) and time to a severe flare (HDA1 intravenous: HR (95% CI) 0.6 (0.37 to 0.81), p=0.0028; HDA2 intravenous: HR (95% CI) 0.6 (0.43 to 0.86), p=0.0049) compared with the placebo group. There was no significant difference in the SF-36 PCS score at week 52 in the intravenous belimumab group compared with placebo in either HDA1 intravenous or HDA2 intravenous populations (table 3). The secondary endpoints explored for the HDA1 subcutaneous (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and HDA2 subcutaneous (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) populations were significant compared with the placebo group only for reduction in risk of severe flare (HDA1 subcutaneous: HR (95% CI) 0.52 (0.30 to 0.92), p=0.0254; HDA2 subcutaneous: HR (95% CI) 0.59 (0.37 to 0.94), p=0.0253), and were not significant for reduction in risk of any flare and the proportion of patients who were able to reduce prednisone use by ≥25% from baseline to ≤7.5 mg/day during weeks 40–52 (table 4).
Secondary efficacy endpoint results (HDA1 and HDA2 intravenous subgroups)
Secondary efficacy endpoint results (HDA1 and HDA2 subcutaneous subgroups)
Discussion
This study evaluated the effect of intravenous and subcutaneous belimumab on disease activity in patients with potentially expanded eligibility criteria for belimumab treatment in SLE, including not only patients with a higher degree of disease activity based on three-variable baseline predictors (HDA1 subgroup: SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) but also the ones with two-variable predictors (HDA2 subgroup: SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA). The HDA1 subgroup criteria were originally based on a previous analysis that suggested greater therapeutic benefit of belimumab compared with standard therapy alone in patients with higher disease activity, anti-dsDNA positivity and low complement at baseline.4 However, the results of these post-hoc analyses demonstrate that the primary endpoint of SRI4 response rate at week 52 was significantly greater in the belimumab group compared with the placebo group using both HDA definitions and in both drug formulations assessed. Furthermore, analysis of the individual components of the primary endpoint showed significant improvements in belimumab-treated patients compared with placebo, in both HDA cohorts and for both drug formulations. Response rates were higher in this post-hoc analysis for belimumab versus placebo compared with the intention-to-treat (ITT) population results from the original trials, in which the ITT population was defined as all patients who had received at least one dose of the study treatment. As with the intravenous results, both subgroups in the subcutaneous post-hoc analyses had a numerically higher SRI4 response rate than the ITT population of the BLISS-SC study. Response rates between the HDA1 (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and HDA2 (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA) subgroups were comparable, indicating that patients fulfilling the expanded criteria of the HDA2 subgroup benefit from belimumab treatment. A significant improvement in both the SRI4 response at week 76 and a reduction in corticosteroid use between week 40 and week 52 in the intravenous belimumab group versus placebo was demonstrated for the HDA1 intravenous subgroup; both endpoints were non-significant in the ITT population in the BLISS-76 study.1 A significant improvement in the SRI4 response at week 76 was not achieved for the HDA2 intravenous subgroup; however, reduction in corticosteroid use between week 40 and week 52 in the intravenous belimumab group versus placebo was significant for the HDA2 intravenous subgroup.
There was no significant reduction in corticosteroid use in the subcutaneous belimumab group versus placebo for either HDA subcutaneous subgroup. There was also no significant difference in BILAG 1A/2B domain scores in the subcutaneous belimumab group versus placebo for the HDA2 subgroup. However, the results were comparable with those of the HDA1 subcutaneous subgroup as well as the HDA1 and HDA2 intravenous subgroups. The lack of significant differences in the reduction of corticosteroid use and new BILAG 1A/2B domain scores compared with placebo for HDA subcutaneous subgroups could be potentially attributed to the smaller sample size in the placebo arm. Nonetheless, the risk of severe SFI flares was significantly reduced in all belimumab patients compared with placebo patients, irrespective of subgroups (HDA1 and HDA2) or drug formulations. Overall, these results indicate that the HDA2 subgrouping provides comparable results with the more stringent HDA1 criteria across most SRI4 components and secondary endpoints assessed in this study. Any differences in speed of response between the HDA1 and HDA2 subgroups may be more a reflection of serologies than clinical manifestations because anti-dsDNA and low complement are components of the SLEDAI score, and a requirement for both to be positive may explain an earlier response seen with HDA1 versus HDA2.
Previous post-hoc analyses of the BLISS trials support the use of belimumab in patients with HDA. Univariate analysis of the pooled BLISS-52 and BLISS-76 data showed that patients with a SELENA-SLEDAI score ≥10, low complement and anti-dsDNA positivity had a greater response to belimumab treatment than those on standard therapy alone.4 A previous prespecified analysis of SLE disease activity in a group of HDA patients from the BLISS-SC study showed a significant improvement in the SRI4 response rate, decreased severe flare incidence and reduced corticosteroid use versus placebo.8 The authors of the BLISS-SC post-hoc analysis used slightly different inclusion criteria, requiring SELENA-SLEDAI score ≥8, compared with a score ≥10 in this study. A previous study that indirectly compared two HDA subgroups in both the intravenous and subcutaneous BLISS trials also found that belimumab treatment had a similar efficacy across administration methods and irrespective of the following HDA criteria: low complement (C3 <0.9 g/L or C4 <0.16 g/L) and anti-dsDNA-positive (≥30 IU/mL) or SELENA-SLEDAI score ≥10.15 This finding suggests that both serological and clinical criteria for HDA are indicative of patients being able to benefit from treatment with belimumab.
An observational cohort study investigating belimumab effectiveness in the real-world setting provided evidence that patients with HDA (SELENA-SLEDAI score ≥10, low complement, anti-dsDNA-positive or a prednisone dose >7.5 mg/day) had reduced disease activity after at least 6 months of treatment with belimumab.16 Another observational study in patients with positive anti-dsDNA antibodies and low complement observed that belimumab treatment decreased disease activity during the study.17
The post-hoc nature of this analysis introduced some limitations to the study. It was not powered to detect statistically significant differences within the included subgroups, and the 2:1 randomisation in the subcutaneous study meant the sample size for the subgroups in the placebo group was small. The smaller sample size of the subcutaneous study in the two subgroups also limits direct comparison between the two routes of administration (intravenous vs subcutaneous). Despite these limitations, this post-hoc analysis provides valuable information on extending belimumab treatment to a wider patient population with a high degree of disease activity.
Conclusion
Belimumab shows improved efficacy, including reduced risk of severe SLE flares, compared with placebo in patients with SLE and HDA for both intravenous and subcutaneous administrations. The present analysis shows clinically meaningful efficacy with belimumab using three-variable baseline predictors (SELENA-SLEDAI score ≥10, low complement and positive anti-dsDNA) and two-variable baseline predictors (SELENA-SLEDAI score ≥10, low complement and/or positive anti-dsDNA), suggesting that expanding the current criteria to include patients with SLE with only two-variable as well as three-variable baseline predictors used in the UK would allow a broader population of patients to be prescribed SLE treatment from which they would derive clinical benefit.
Acknowledgments
We would like to thank Milena Kurtinecz and Mark Tumelty of the statistics department of GSK Research and Development for their help with statistical analyses. Medical writing support was provided by Olga Conn, PhD, of Fishawack Indicia, UK, and was funded by GSK.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Lupusdoc
Contributors TM, IB, DDC, MI, DB and DR contributed to conception or design of the study. DDC contributed to acquisition of data. TM, IB, DDC, DB and CW contributed to data analysis or interpretation. All authors meet the requirements for authorship, have contributed towards the preparation of the manuscript, have approved the final submitted version and have agreed to be included as authors.
Funding This study (GSK Study 207878) was funded by GSK.
Disclaimer The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests TM, MI, DB, CW and DR are employees of GSK and hold stocks and shares in the company. IB is a National Institute for Health Research (NIHR) senior investigator and is funded by Versus Arthritis and the NIHR Manchester Biomedical Research Centre. IB has also received speaker’s bureau and advisory board grants from UCB, has participated in advisory boards and steering committees for AstraZeneca, is a member of Independent Data Safety Boards for Medimmune and MS, has received grants from Genzyme Sanofi, and has participated in advisory boards for Eli Lilly. DDC reports participation in advisory boards and consultancies for GSK, Human Genome Sciences, Eli Lilly and Roche, and has received consulting fees and/or has participated in clinical trials for GSK, Bristol-Myers Squibb, TEVA, Merck Serono and Eli Lilly.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Anonymised individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.