Article Text

Abstract
Objective Two apolipoprotein L1 (APOL1) risk variants (RV) are enriched in sub-Saharan African populations due to conferred resistance to Trypanosoma brucei. These variants associate with adverse renal outcomes by multiple causes including SLE. Despite emerging reports that SLE is common in Ghana, where APOL1 variant allelic frequencies are high, the regional contribution to SLE outcomes has not been described. Accordingly, this prospective longitudinal cohort study tested the associations between APOL1 high-risk genotypes and kidney outcomes, organ damage accrual and death in 100 Ghanaian patients with SLE.
Methods This was a prospective cohort study of 100 SLE outpatients who sought care at Korle bu Teaching Hospital in Accra, Ghana. Adult patients who met 4 American College of Rheumatology criteria for SLE were genotyped for APOL1 and followed longitudinally for SLE activity as measured by the Safety of Estrogens in Lupus National Assessment-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) hybrid and organ injury as measured by the Systemic Lupus International Collaborating Clinics Damage Index (SDI) at baseline and every 6 months for 1 year. Outcomes of interest were kidney function, SDI and case fatality.
Results Assuming a recessive inheritance, the APOL1 high-risk genotype (2RV) associated with end-stage renal disease (ESRD) at an OR of 14 (p=0.008). These patients accrued more SDI points particularly in renal and neurological domains. The SDI was 81.3% higher in 2RV patients compared with 0RV or 1RV patients despite no difference in SLE activity (p=0.01). After a 12-month period of observation, 3/12 (25%) of the 2RV patients died compared with 2/88 (2.3%) of the 0RV or 1RV carriers (OR=13.6, p=0.01). Deaths were due to end-stage kidney disease and heart failure.
Conclusion APOL1 RVs were heritable risk factors for morbidity and mortality in this Ghanaian SLE cohort. Despite no appreciable differences in SLE activity, APOL1 high-risk patients exhibited progressive renal disease, organ damage accrual and a 13-fold higher case fatality.
- lupus erythematosus
- systemic
- lupus nephritis
- polymorphism
- genetic
- cardiovascular diseases
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
APOL1 high-risk genotype is known to associate with SLE nephritis progression.
In this cohort, APOL1 high-risk genotype contributed to SLE damage accrual.
APOL1 high-risk genotype carriers exhibited excess damage despite no differences in SLE disease activity.
Case fatality rate was 14 fold higher in APOL1 high-risk genotype SLE patients.
APOL1 genotype may be an important genetic contributor to SLE morbidity and mortality.
Introduction
Two coding change variants in the apolipoprotein L1 (APOL1) gene, G1 (S342G and I384M) and G2 (N388del;Y389del), have recently been identified in sub-Saharan African genomes. Some of the highest reported APOL1 variant allele frequencies can be found in Ghana, where up to 41% and 13% of alleles carry the G1 and G2 variants, respectively.1 The variants are thought to have been evolutionarily conserved due to a protection against Trypanosoma brucei, the parasite cause of African trypanosomiasis.1 This infectious protection comes with a higher risk of several adverse phenotypes, including end-stage renal disease (ESRD) by multiple causes and SLE nephritis.2–4 Several epidemiological studies on the African continent have linked APOL1 to progressive renal disease, hypertension and stroke; however, no study has been performed in SLE.5–9
SLE may represent an important context under which APOL1 variants confer heightened risk. Associations between adverse phenotypes and APOL1 high-risk genotypes, defined as two variants in any combination (G1/G1, G1/G2, or G2/G2), are highly heterogeneous. For example, the odds of developing chronic kidney disease range from 1.5 to 2.0 in otherwise healthy individuals, to 2.5–7.3 in SLE nephritis, to 29–80 in HIV-associated nephropathy.10–12 In SLE, the APOL1 high-risk genotype has been shown to associate with collapsing glomerulopathy and progression to ESRD.2 12 Even carrying a single variant copy has been associated with hypertension, prevalent cardiovascular disease and kidney injury in SLE.13 14 One hypothesis for this is that gene penetrance is contingent on environmental ‘second hits’.15
Consistent with its immune function, APOL1 expression is highly responsive to inflammatory signals.16 We and others have shown that a wide range of stimuli, including endogenous and exogenous interferons, autoantigens and SLE serum, increase APOL1 expression and therefore intracellular accumulation.16 17 APOL1 contains both a BH3 domain that participates in the cellular stress response through autophagy, and a pore-forming domain that can traverse phospholipid bilayers in a pH-dependent fashion.18 19 At high expression levels, intracellularly accumulated APOL1 shifts from a pro-autophagic function towards a cytotoxic pore-forming function.18 20 Pore formation has been shown to cause injury in kidney and vascular-related cells including podocytes and endothelial cells.21
Recent literature reports that severe SLE is more common in Ghana than previously appreciated. For example, a retrospective review showed that SLE represented 5.28/1000 inpatient admissions at Korle bu Teaching Hospital in Accra, Ghana.22 These patients experienced high mortality as 43% died, most commonly of kidney disease, over a 2-year period.23 It is unclear how APOL1 high-risk genotype contributes to this trend. We therefore conducted a prospective cohort study comparing outcomes across APOL1 genotype groups in patients with SLE seeking care at Korle bu Teaching Hospital in Accra, Ghana.
Methods
Study population
Enrolment was initiated at Korle bu Teaching Hospital outpatient rheumatology clinics between 2015 and 2017. A consecutive sample of 100 individuals seeking clinical rheumatology care was invited to participate. Inclusion criteria were as follows: (1) ≥18 years of age, (2) self-reported Ghanaian ancestry and (3) fulfilling at least four 1982 American College of Rheumatology (ACR) criteria for SLE.24 Patients who were unwilling or unable to sign consent were excluded.
Patient involvement
Patients were involved in the conduct of this research study. The study coordinator team in Accra, Ghana included patient advocates trained to engage and inform other patients participating in The Rheumatology Initiative support group, founded by author, DD. During the study, interactions between the study team and patients were facilitated through monthly educational sessions. Once the study is published, patients and participants will be informed of the results through these support group sessions.
Data collection
Initial enrolment: each study participant completed a survey to assess demographics, medication lists and ACR SLE criteria. Both the Systemic Lupus International Collaborating Clinics (SLICC) Damage Index (SDI)25 and SELENA-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) hybrid26 27 were recorded during routine clinical visits. The SELENA-SLEDAI hybrid28 comprises the same definitions as the original SELENA-SLENA-SLEDAI,29 but uses the definition of the SLEDAI and SLEDAI-2K27 to score the domain of proteinuria. We chose the hybrid because the SELENA-SLEDAI only scores 4 points for proteinuria when there is an increase of 500 mg/24 hours over the previous visit. For studying this cohort, we favoured capturing ongoing proteinuria as defined by the SLEDAI and SLEDAI-2K, both of which score proteinuria of >500 mg/24 hours regardless of the previous visit. Complete physical examination, vital signs and anthropometric measurements were recorded. SLE serological results including anti-ANA, antidouble-stranded DNA antibodies (dsDNA), anti-SSA/Ro antibodies (Ro), anti-SSB/La antibodies (La), anti-Smith antibodies (Smith), anti-U1/RNP antibodies (RNP) and antiphospholipid antibodies were recorded. Baseline urine samples were evaluated by dipstick and urinalysis where available (Acon Laboratories, San Diego, California, USA), and saliva and whole blood were collected for DNA extraction (Oragene saliva collection kits DNA Genotek, Ottawa, Ontario, Canada) and serum and plasma (Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA), respectively. Serum, plasma and saliva samples were dated, batched and shipped to the New York University (NYU) laboratory for processing.
Follow-up: each patient was followed longitudinally at 6-month intervals for 1 year. During follow-up visits, physical exams, SELENA-SLEDAI, laboratory review and medication lists were recorded. Serum, plasma and urine dipstick were again taken or recorded.
Sample assessment
Apolipoprotein L1 genotyping: study patients’ genomic DNA was isolated from saliva using Oragene reagents according to the manufacturer’s instructions (DNA Genotek). As described previously, DNA isolates were stored at −80°C, and quantitated using a Nanodrop-1000 spectrophotometer (Nanodrop Products, Wilmington, Delaware, USA). DNA templates (100 ng) were used for conventional PCR as previously described.14 A single 300-base pair DNA segment containing the APOL1 gene, including reference G0 allele and polymorphisms G1 (rs73885319 and rs60910145) and G2 (rs71785313), was amplified using ApliTaq Gold 360 DNA Polymerase (Applied Biosystems, Foster City, California, USA). For quality control, DNA was elongated in both forward and reverse directions. Genotypes were analysed using the Genewiz online platform as previously described.14
Autoantibody serological screening: batched and shipped serum samples were screened for ANAs using the BioPlex 220 ANA Screen in the NYU Langone Hospitals clinical lab. This automated system uses multiplex technology to measure 13 antibodies including SLE-relevant antigens dsDNA, chromatin, RNP-68 kDa, SSA-52 kDa, SSA-60 kDa and Sm/RNP as described.30 A mixture of antigen-coated beads was combined with patient sample and diluent for an incubation period of 20 min at 37°C. Beads were washed and treated with antihuman IgG antibody conjugated to phycoerthrin dye for a 10 min incubation. Excess conjugate was removed, and the mixture was passed through a detector. The bead type and amount detected were read and reported.
Statistical analysis
To test the association between APOL1 genotype and SLE outcomes, we treated APOL1 genotype as the predictive variable (coded Number_Alleles) and composite SDI, renal function and case fatality as outcome variables (coded SLICC, Renal and Mort, respectively). The associations between primary predictor and outcome variables were tested using Poisson regression for the SLICC damage and generalised estimation equations (GEE) to identify factors associated with the composite renal function measure overtime. The distribution of sex, age, SELENA-SLEDAI score, disease duration, kidney function and SLE criteria were assessed across genotype. Factors that associated with the outcome variables at a significance level <0.10 were treated as co-variates; they included the clinical SELENA-SLEDAI score, disease duration and a renal function composite score obtained by performing a factor analysis of mean arterial pressure (MAP), estimated glomerular filtration rate (eGFR) and proteinuria on urine dipstick followed by a varimax rotation.31 32 The factor analysis was performed using data from the first visit. The eigenvector and the rotation matrix from this analysis was then used to derive the composite scores at the second and third visit.
Complete case and analyses are presented as the primary result; however, given the high rates of missing data, especially at the third visit, results from analyses using multiple imputations are also presented. Multiple imputation was performed assuming missing at random and monotone missingness. Results from the imputed datasets were combined using Rubin’s approach as implemented in the SAS V.9.4 MIANALYZE procedure.33 34 Analyses were performed using R 3.6.3 or SAS V.9.4.
Results
Characteristics of the cohort
We enrolled 100 outpatients who met inclusion criteria and were consented to participate at Korle bu Teaching Hospital in Accra, Ghana during their routine clinical care. Demographic characteristics are shown in table 1. The mean±SD age was 32±9.4 years and 99% of the patients self-identified as female. The most common ethnic groups were Akan (42%) followed by Ewe (28%) and Ga (20%) representing 90% of the study participants.
Characteristics of the study population
On average, patients had a disease duration of 2.3±2.3 years at the baseline. The reported time from symptom development to diagnosis was 8.5±15.8 months. The most common ACR criteria was arthritis (77%), followed by lupus nephritis (53%), serositis (50%) and haematological disorder (50%; table 1). Of the cohort, 76% had ANAs completed for the purposes of clinical care with a median titre of 1:320 and a range from 1:40 to 1:5120. The remaining 24% were diagnosed clinically. On testing sera in the NYU clinical laboratory, 93% of patients were ANA positive. The most common autoantibodies were RNP, Smith, dsDNA and Ro, which were positive in 65%, 56%, 56% and 54% of the cohort, respectively (table 1). The most commonly used medications were glucocorticoids (92%) and hydroxychloroquine (87%). Non-steroidal anti-inflammatory drug (NSAID) use was also common with 73% of the cohort taking these drugs at baseline survey. The most common disease-modifying antirheumatic drugs (DMARDs) were azathioprine, cyclophosphamide and methotrexate with 37%, 13% and 10% of patients receiving these drugs.
APOL1 genotype associates with SLICC Damage Index
The average SDI) was 1.1±1.5 (mean±SD) at enrollment; 54% of the cohort had a non-zero SDI with on average 2.3 years of SLE disease duration on enrolment. The frequencies of the ancestral G0 and risk variant (RV) G1 or G2 allele were 0.64, 0.21 and 0.12, respectively, and the allele distribution did not deviate from Hardy Weinberg Equilibrium (p=0.2, table 2).
Cohort patients ethnicities and APOL1 allelic frequencies
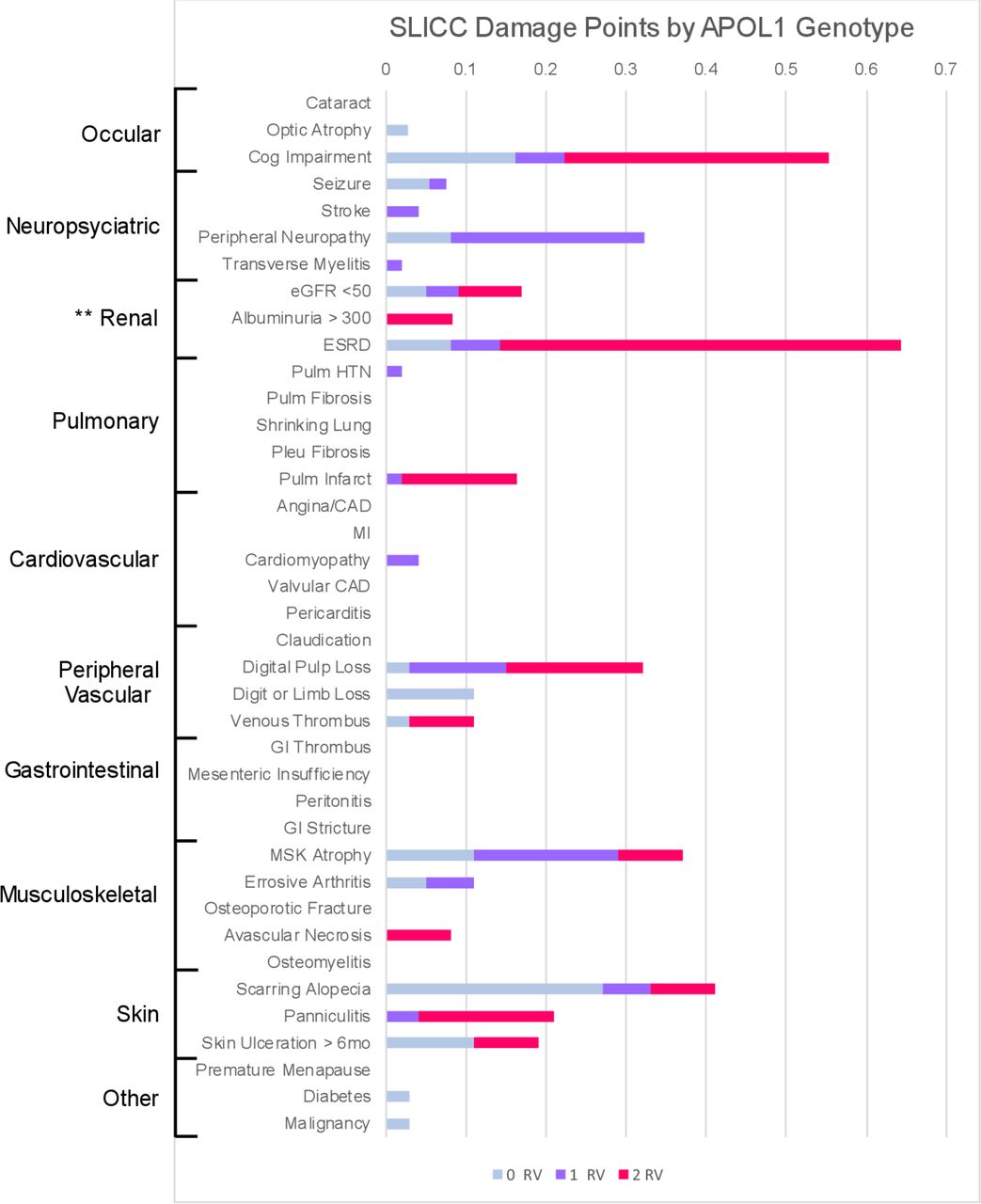
APOL1 high-risk (2RV) patients exhibited numerically higher SDI, compared with patients with zero or one variant, although this did not reach statistical significance (mean SDI 0RV or 1RV=0.99±1.2 vs 2RV=1.75±2.6, p=0.1). APOL1 high-risk patients accrued more SDI points in renal, cardiac and neurological domains (figure 1). To model SDI points accrued by genotype, a Poisson regression with a logarithmic link was used (table 3). In the unadjusted model, 2RV patients showed an 81.3% (95% CI 12.3 to 192.7) higher SDI compared with APOL1 low-risk (1RV or 0RV) patients. In the first model adjusted for average SLEDAI score, the increase was 72.2% (95% CI 6.5 to 178.3) among 2RV patients, relative to 0RV or 1RV patients. An increase of one unit in the 12-month average SLEDAI score corresponded to a 4.7% (95% CI 2.0 to 7.3%) increase in the SLICC damage score (table 3). Further adjustment for disease duration in the third model did not change this result (1.5%, 95% CI −6.5 to 10.2). Unadjusted and adjusted model summaries including both complete case data and data with imputed values are shown in table 3.
APOL1 genotype associates with SLICC Damage Index
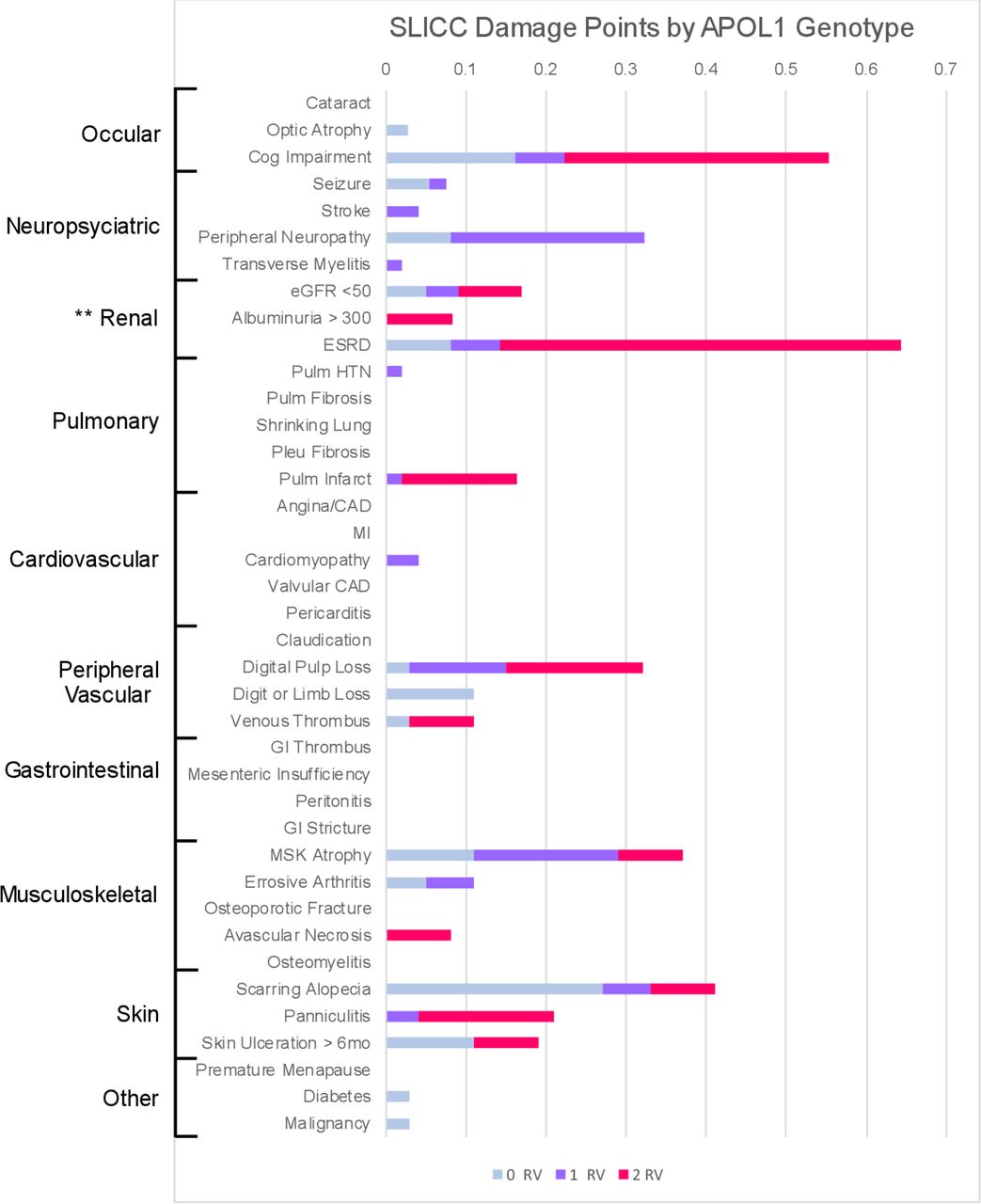
SLICC Damage Index (SDI) across APOL1 genotype: stacked bar chart visualising the mean SDI points accrued by genotype group (0RV in blue, 1RV in orange, 2RV in grey). Each damage criterion is ordered by affected organ system (shown to the left). APOL1, apolipoprotein L1; RV, risk variant.
APOL1 genotype is associated with renal function
A higher proportion of 2RV patients had 2+ or greater proteinuria scores on urine dipstick at baseline, 6 months and 12 months (figure 2A). These patients also exhibited higher MAP at each time point, and lower eGFR as measured by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula (p=0.01 for each outcome MAP and eGFR; figure 2B and C). During the 12-month period, of the five patients who progressed to ESRD as defined by eGFR ≤15 mL/min/1.73 m2 or referral for haemodialysis, three were 2RV carriers. Assuming a recessive mode of inheritance, carrying 2RV in APOL1 is associated with developing ESRD (OR=14 (95% CI 2.1 to 95.2); p=0.008).

Distribution of kidney function parameters by APOL1 genotype and time point. (A) The bars represent the proportion of patients in each urine dipstick range (0+−3–4+). The x-axis represents percentages, and the y-axis represents the genotype groups by time point (0M=month zero, 6M=month 6, 12M=month 12). At each time point, the proportion of patients in the higher dipstick propteinuria ranges was higher in the 2RV patients. These differences did not reach statistical significance. (B) The box plots represent the distribution of mean arterial pressure (mm Hg) values by genotype groups and time point. Overall, 2RV patients exhibited significantly higher mean arterial pressure values (p=0.01). (C) The box plots represent the distribution of mean eGFR (mL/min) values by genotype groups and time point. Overall, 2RV patients exhibited significantly lower eGFR values (p=0.01). **P=0.01. APOL1, apolipoprotein L1; eGFR, estimated glomerular filtration rate; RV, risk variant.
To capture the effect of 2RV status on urine dipstick proteinuria, MAP and lower eGFR, a weighted average of each variable was created to fully represent kidney function. This weighted average was designated as a composite variable called ‘factor one’, which was proportional to kidney function and computed with the following loadings: factor one=(−0.67×MAP)−(0.28×dipstick protein)+(0.63×eGFR). In the GEE model, APOL1 variants associated with a decrease in the renal factor one (−0.76, 95% CI −1.35 to –0.16). On controlling for SDI, this association between APOL1 variants and factor one remained (−0.63, 95% CI −1.1 to –0.16). Lastly, this association was not mitigated by disease duration (table 4).
APOL1 genotype associates with renal function
APOL1 genotype is associated with case fatality
The case fatality rate was significantly higher in 2RV patients. Over 12 months, 2.3% (2 out of 88) patients with 0RV or 1RV died compared with 25% (3 out of 12) 2RV patients corresponding with an OR of 13.6 (95% CI 1.4 to 182.6). Deaths in the 0RV or 1RV group were due to ESRD and sepsis during pregnancy. In the 2RV patients, deaths were due to ESRD and heart failure. Case fatality rate and causes of death are summarised in figure 3. The association between the APOL1 high-risk genotype and case fatality remained on controlling for average SLEDAI at an OR of 14.8 (95% CI 2.0 to 109.4). Taken together, these data suggest that APOL1 high-risk genotype is a contributor to SLE mortality independent of disease activity.

{kind=link}
{kind=link}
{kind=link}
Case fatality rate by APOL1 genotype. (A) Bars represent each genotype group (0RV, 1RV or 2RV). The y-axis represents the percentage of patients in that genotype group who died, and the x-axis shows the study time point. **P<0.01. (B) Table represents the causes of death at each time point by genotype group. APOL1, apolipoprotein L1; ESRD, end-stage renal disease; Pulm, pulmonary; RV, risk variant.
Discussion
The central findings in our study were that carrying 2RV was associated with proteinuria, higher MAP and lower average eGFR levels despite similar SLE activity measures. Compared with the 0RV and 1RV patients, the 2RV group accumulated more points on the SDI both in renal domains and in those suggesting microvascular disease including cognitive impairment, digital pulp loss and panniculitis. The case fatality rate in 2RV carriers was 13.6-fold that of the 0RV or 1RV carriers on controlling for SLE disease activity. Deaths in the 2RV patients were due to ESRD and heart failure, consistent with previously identified APOL1 variant-associated phenotypes. Importantly, the APOL1 risk genotype correlated with SDI, renal progression and case fatality independent of SLE disease activity, suggesting that in Ghanaian patients with SLE, these variants may be significant prognostic indicators.
Associations between the APOL1 high-risk genotype and non-diabetic kidney disease by multiple causes have been consistent with recent reports of early onset renal disease in West Africa.2 3 6 10 35 36 Notably, reports from Ghana indicate that the APOL1 high-risk genotype is associated with small vessel ischaemic stroke, a risk factor for cognitive impairment.9 In the USA, reports implicate the APOL1 high-risk genotype in extrarenal phenotypes including pre-eclampsia and non-diabetic microvascular stroke.37–39 The current study adds to the understanding of APOL1 high-risk genotype manifestations in SLE. In addition to renal domains, 2RV patients accrued more damage due to cognitive impairment, pulmonary infarction and digital pulp loss.
SLE is an extremely heterogeneous disease, with variation in clinical manifestations, race and ethnicity, disease onset, medication requirements and disease activity all contributing to morbidity and mortality.25 40 41 Patients with SLE of African ancestry have been shown to accrue more organ damage, and suffer with more comorbidities than patients of European ancestry.41 42 These differences are multifactorial and thought to be largely driven by socioeconomic factors, however genetics may also play a role.43–45 Genetic susceptibility to early damage accrual, particularly in renal and cardiovascular domains, is implicated in early SLE deaths.46 Recently, a Swedish group found that a higher cumulative genetic risk score spanning 57 SLE-associated loci was implicated in SLE onset, severity, organ damage and death; however, APOL1 was not identified likely due to a lack of patients with African continental ancestry.46 The current study highlights the need to perform additional genetic susceptibility analyses in patients with SLE of diverse ancestral backgrounds.
Several sub-Saharan African case series have identified high SLE-related morbidity and mortality due to infections and cardiorenal disease.22 23 47 While epidemiological studies on the African continent are sparse, individuals of recent African ancestry throughout the diaspora have some of the highest reported incidence and prevalence of SLE.48 49 This trend is compounded by higher morbidity and mortality due to renal, neurological and cardiovascular disease.40 41 48 49 In the USA, SLE ranked among the top 10 causes of death in African-American women from the second through fifth decades of life.50 Moreover, African-American patients with SLE have demonstrably higher prevalent comorbid hypertensive, renal and cardiovascular disease compared with their European American counterparts.42 Damage accrual on renal and cardiovascular domains of the SDI are independent risk factors for early SLE mortality.25 While many socioeconomic factors are at play, the observed association between these critical outcomes and APOL1 risk genotype in SLE may be an underappreciated genetic contribution.
The current study is not without limitations. Given SLE disease heterogeneity, other genetic, treatment and environmental factors contribute to damage accrual, kidney disease susceptibility and mortality. This highlights the need for genome-wide association studies in a continental African SLE population. The generalisability of the findings is limited by the small sample size with 12% of participants in the 2RV group. Larger studies are necessary to better understand the associations between the APOL1 polymorphisms and systemic disease. Patients were often unable to afford or obtain laboratory studies at each visit, thus limiting longitudinal laboratory data. This could have underestimated SLE activity based on available disease measures.51 In addition, high 1-year mortality could be attributed to limited access to DMARDs and life-saving dialysis treatments. Study patients commonly used NSAIDs and corticosteroids both of which have adverse cardiorenal effects. However, this phenomenon influenced all study patients across genotype groups. Despite these limitations, this novel sub-Saharan African study offers important information regarding APOL1 risk phenotypes in SLE.
Conclusion
These data implicate APOL1 high-risk genotype in SLE renal progression, extrarenal damage accrual and mortality in this Ghanaian SLE cohort. Furthermore, we identify a potential genetic associations with high SLE-associated mortality in sub-Saharan Africa. Additional research is required to determine the utility and cost-effectiveness of APOL1 genotyping in patients with SLE of recent African ancestry for risk stratification and disease prevention.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
All study participants signed informed consent prior to participating in this study. Ethics approval was granted by The University of Ghana Ethics Board. This study was conducted in compliance with the Declaration of Helsinki.
Acknowledgments
The authors would like to thank the Buyon/Clancy laboratory where genotyping work was completed for this project, and the advice of mentors and peers including Drs Jill Buyon, Robert Clancy, Timothy Niewold, Greg Silverman, Janina Jeff and Peter Izmirly. The authors would also like to thank the patients who participated in this study, and The Rheumatology Initiative that allowed the team to collaborate with, educate and support patients with SLE in Accra, Ghana.
References
Footnotes
Twitter @Ashira_MD
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors Each author made substantial contributions to conception (AB, IDD, JN), data acquisition and analysis (AB, MR, FA, JD) and/or drafting the work (AB, HA, TA). In addition, all authors have approved the submitted version of this manuscript, and have agreed to be personally accountable for his or her own contributions.
Funding This project was supported in part by the Rheumatology Research Foundation Scientist Development Award to AB; the National Centre for the Advancement of Translational Science (NCATS) (KL2 1UL1TR001445) to AB and The Judith & Stewart Colton Centre for Autoimmunity Scholar Award to AB.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the 'Methods' section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.