Article Text

Abstract
Introduction As chronic systemic autoimmune disease, which can affect every organ, SLE is creating significant burden and increased mortality. Despite better outcomes over the past decades by optimising standard of care, new interventions are needed for further improvements. Changing strategy to ‘treat-to-target’ (T2T) may be a promising concept proven successful in other chronic diseases.
Methods and analysis In this cluster-randomised trial, SLE centres will be assigned 1:1:1 to standard of care (SoC), remission (no clinical disease activity+prednisolone ≤5 mg/day+Physician Global Assessment (PGA 0–3) <0.5±immunomodulatory treatment) or and Lupus Low Disease Activity State (LLDAS, low disease activity+prednisolone ≤7.5 mg/day+PGA ≤1+no new disease activity). Per arm, 424 patients will be included. Intervention centres receive a standardised training on T2T and shared decision-making (SDM). In intervention centres, patients not in target enter a phase of tight control with six weekly visits and treatment adjustments (at least four visits) or until the target is reached and maintained. Patients in target are reassessed every 12 weeks. In case of flare, they can enter tight control based on SDM. In the SoC arm, patients receive their usual three to six monthly controls and treatment adjustments according to the physician’s discretion. Study duration is 120 weeks using change in damage and health-related quality of life (HRQoL) as major outcomes. The primary endpoint will be damage accrual at 120 weeks as measured by the Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index and will by analysed by a mixed model.
Conclusions This is the first trial to assess if the implementation of a T2T concept in clinical care minimises damage accrual and improves HRQoL in patients with SLE. Comparison of remission and LLDAS will help to identify the target with the best benefit–risk ratio concerning attainability, adverse events and damage. The emphasis on SDM will strengthen patient autonomy and will improve both their satisfaction and medical condition.
- lupus erythematosus
- systemic
- outcome assessment
- healthcare
- quality of life
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Due to earlier diagnosis and optimised use of approved drugs, the prognosis of patients with SLE has improved over the last four decades.1 Although SLE can, in some cases, still be life-threatening, the general disease course changed to a chronic disease with damage accrual as the major risk factor for reduced health-related quality of life (HRQoL) and premature mortality.2 3 Better control of disease activity and thus reduction of damage accrual either by novel, more efficient drugs or new treatment strategies are urgently needed.
Treat-to-target (T2T) has become an accepted effective concept in several disciplines, such as diabetes and cardiovascular risk management and recently in rheumatology.4 5 Pursuing a target in rheumatoid arthritis (RA) such as low disease activity (LDA) or remission yields significant increases in the rate of remission, improves physical function, reduces radiographic progression and optimises HRQoL.6 7 To establish a T2T strategy, three conditions must be met: first, a suitable target has to be defined. Second and third, therapeutic options to reach the target and outcome measures to monitor target achievement are necessary. All requirements are now fulfilled in SLE. With DORIS remission and Lupus Low Disease Activity State (LLDAS), internationally accepted definitions were developed.8 9 Cohort evaluations confirmed the face validity of remission and LLDAS, as being in one of the states was significantly associated with improved HRQoL and less damage accrual.10–12 A shorter time in remission than in LLDAS was required to significantly reduce damage progression.13 Post-hoc analyses of clinical trials also indicate that LLDAS can generally be achieved in 5.8%–12.5% of patients with active SLE after 52 weeks.14 The missing piece of evidence is now to show that bringing patients into target (‘treating to target’) in clinical care has similar beneficial consequences as being in target. A randomised controlled trial is highly needed to confirm the benefit of tight control and targeting patients to remission/LLDAS, and assess its requirements and its immediate impact on disease outcome. Further, it could facilitate the early identification of patients in need for a therapeutic change.
Shared decision-making (SDM) describes a process by which physicians and patients contribute equally to the decision-making process and agree on treatment decisions. The aim is to provide high-quality care based on scientific evidence and clinical experience while integrating patients’ values and preferences,15 thus promote patient autonomy. Since informed consent is at the core of SDM, patients need to fully understand the advantages and disadvantages of all treatment options.16 The integration of SDM into clinical care of patients with SLE has been incorporated into the current European League Against Rheumatism (EULAR) recommendations for the management of SLE.17 However, specific recommendations on how to implement SDM into clinical care do not exist.
This will be the first randomised controlled trial in SLE using a tight-control T2T approach to achieve either remission or LLDAS in clinical care. T2T (including SDM) has never been investigated as strategy in such a complex disease before; this approach is particularly unique by including concomitant therapy (glucocorticoids (GC) dosage) with impact on damage as part of the target. At the time of submission, LUPUS-BEST is in preparation phase.
Methods and analysis
Study overview
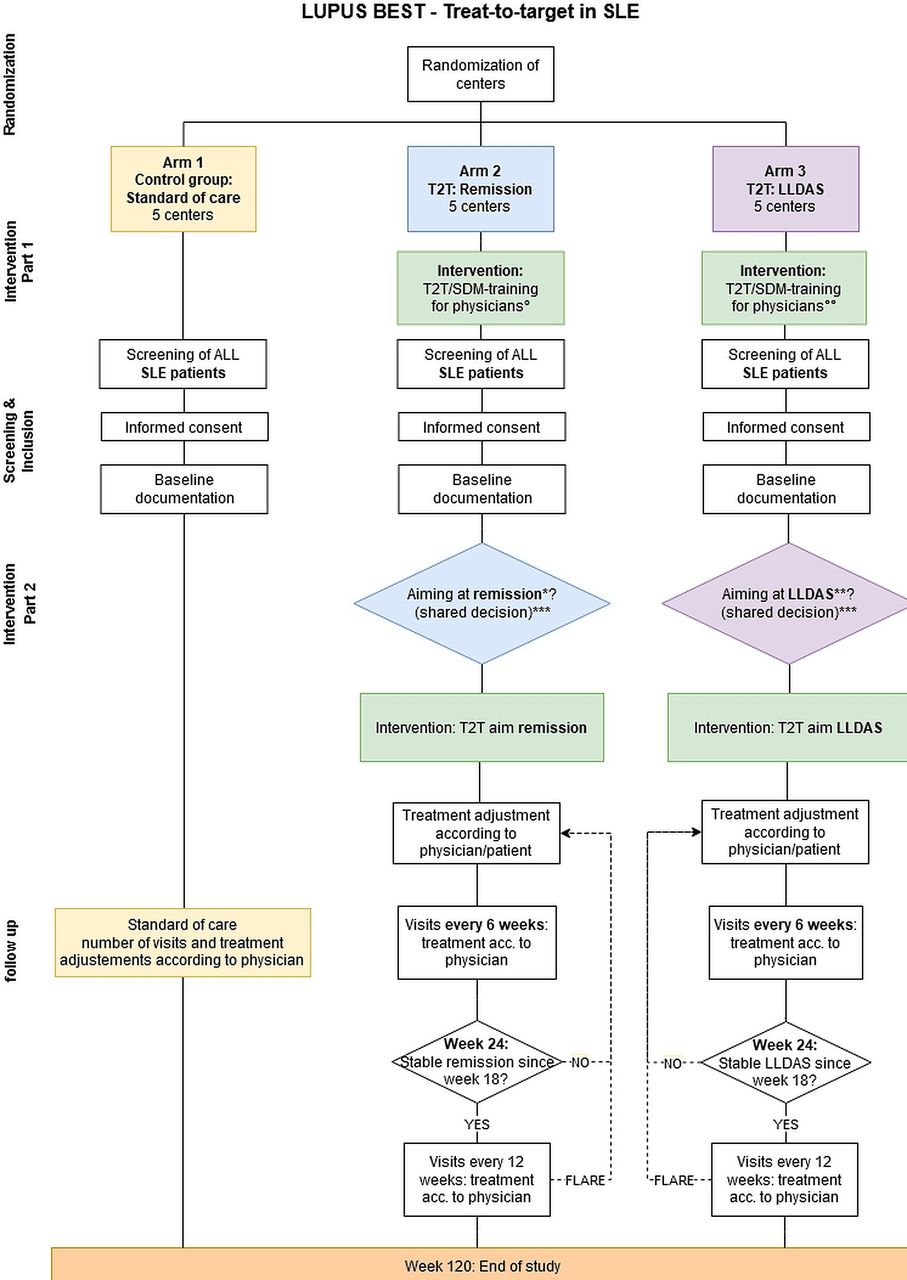
LUPUS-BEST is a prospective, multicentre, cluster-randomised trial that will enrol subjects across 15 study centres in Germany over a 4-year period. A schematic representation of the study design is presented in figure 1. The trial uses a cluster-randomised design, where the study centres will be randomly assigned 1:1:1 to either remission, LLDAS or standard of care (SoC). Patients with SLE above the age of 18 years will be screened end enrolled when eligible. With tight control and frequent treatment evaluations based on SDM, patients in the intervention arms will be treated to target and the primary outcome will be assessed at week 120, the end of the trial.

Trial flow chart. °Remission training; °°LLDAS training. *Remission: cSLEDAI-2K=0 and GC ≤5 mg/day and PGA <0.5; ±immunomodulatory therapy. **LLDAS: SLEDAI-2K ≤4 and GC ≤7.5 mg/day and PGA ≤1 and no new disease activity. ***Documentation of reason for T2T refusal. GC glucocorticoids; LLDAS Lupus Low Disease Activity State; PGA, Physician Global Assessment; SDM, shared decision-making; SLEDAI-2K, SLE Disease Activity Index 2000; T2T, treat-to-target.
The trial will primarily be conducted on a national level, however, international extension would be encouraged. Public funding has been applied for. In addition, contact to pharmaceutical sponsors has been established in order to sort out unrestricted financial support.
Patient population and eligibility
The trial is accessible to all patients with SLE in the participating centres classified according to validated criteria (EULAR/American College of Rheumatology (ACR) criteria,18 Systemic Lupus International Collaborating Clinics (SLICC) criteria19), above the age of 18 years, with written informed consent and sufficient German language skills, regardless of their disease manifestations to represent the broad variety of SLE manifestations and real-life data. Exclusion criteria are life-threatening SLE manifestations that require intensive care treatment; active life-threatening diseases other than SLE; and active malignancies as well as active, chronic or recurrent viral infection that, based on the investigators’ clinical assessment, make the subject an unsuitable candidate for the study, including hepatitis, HIV and tuberculosis. In case of pregnancy or breast feeding during the trial, the intervention will set on hold if recommended and the patient will be documented according to the trial protocol. These criteria allow for inclusion of the vast majority of patients with SLE treated in the participating centres and the study population will thus reflect and represent the whole spectrum of patients with SLE in real life in Germany. Patients of all genders and >18 years of age will be included, hence generalisability is expected to the majority of adult patients with SLE.
Interventions
T2T in SLE is defined by tight control in terms of frequent visits and treatment optimisation based on SDM aiming for patients with SLE to reach remission on treatment (arm 2) or a state of LDA (LLDAS, arm 3). Remission is defined as the lack of clinical disease activity, a steroid dosage of ≤5 mg prednisolone per day and a Physician Global Assessment (PGA) of the current disease activity of <0.5 on a numerical rating scale from 0 to 3.8 LLDAS is characterised by LDA with no new disease activity, a steroid dose of ≤7.5 mg prednisolone per day and a PGA ≤1.20
The study personnel in the intervention centres will receive 8 hours of standardised training on T2T and SDM. The training will take place in an interactive group format at each centre and will be performed by an experienced SDM expert and the study organisers who have been treating patients with SLE for 30 years and follow T2T approaches in other rheumatic diseases.
For this purpose, an approved SDM training format21 22 will be combined with established T2T approaches. It will integrate SLE-specific case vignettes and decision support materials (Patient Decision Aids, decision boards, treatment flow charts, risk charts) to be generated according to International Patient Decision Aid Standards criteria23 and in close feedback loops between rheumatologists, SDM experts, patients and patient representatives beforehand.
All patients with SLE in the participating centres will be invited to join the trial. Those enrolled in the study after being screened for eligibility and giving informed consent will be documented in a standardised manner at every visit during the total trial period of 120 weeks. Approved medications consist of antimalarials as basic therapy, GC and/or immune-modulating agents like methotrexate, azathioprine, mycophenolate, cyclophosphamide and/or belimumab as well as rituximab. Treatment decisions are made according to the physicians’ discretion and SDM based on Good Clinical Practice and may also include other drugs.
In the intervention centres, patients will be assessed for being in target at every visit. Patients already in target at the screening visit will be documented in a standardised manner in 12 weekly assessments if still in target at the baseline visit thus skipping the T2T loop and entering the follow-up phase at once. Patients who do not meet remission/LLDAS should receive a treatment adjustment based on SDM: (a) modification of the current immunomodulator(s) and/or (b) addition of a new immunomodulator and/or (c) adjustment of the current GC dosage. In case of GC adaption, a GC tapering scheme will be assigned for the upcoming 6 weeks.
Depending on the clinical situation and based on SDM, the target (remission/LLDAS) can be addressed by one or more tight-control loop(s). As exception from this standardised process, the option of no treatment adjustment is available but must be documented and explained. In case of high PGA as the only ‘non-targetable’ item, which encapsulates the overall disease activity, the reason(s) for high PGA must be documented and addressed.
All patients, who do not meet remission/LLDAS criteria and thus receive a treatment adjustment, will be reassessed every 6 weeks and treatment may be modified at every visit based on SDM. Tapering or dose increase schemes may be applied and lead to dosing modifications in-between the visits.
After 24 weeks of tight control, patients fulfilling remission/LLDAS for at least 6 weeks since the last visit will change to 12 weekly assessments. If remission/LLDAS is not achieved at week 24, tight control continues until (a) the target is achieved in two consecutive visits (with 6-week interval), (b) the end of study after 120 weeks, or (c) physician and patient agree to refuse T2T. If patients fulfilling remission/LLDAS flare as measured by the SLE Disease Activity Index Flare Index or miss the target at any time during follow-up, they will be invited to reassign to tight control and T2T for another 24-week loop. If T2T is refused, the patient will receive 12 weekly assessments with the option to start tight control any time later in case of agreement.
Patients in the LLDAS arm, who reach their target, do not have to be retained in LLDAS but can reach remission as well by, that is, further steroid taper or an improvement in PGA. Hence, these patients could reach remission by SoC. However, patients in target in the LLDAS arm should not receive a treatment intensification to reach remission.
Pharmaceutical treatment decisions will be guided by a decision scheme based on the recently updated EULAR recommendations17 and will be taken in accordance with SDM between patients and treating physicians (experienced rheumatologist or rheumatologist in training). In the intervention centres, all handling physicians have to be trained for T2T and SDM. The flexibility of the intervention scheme with the option to leave and re-enter tight control reflects clinical care, and presumably more patients will achieve the aimed target.
Study endpoints
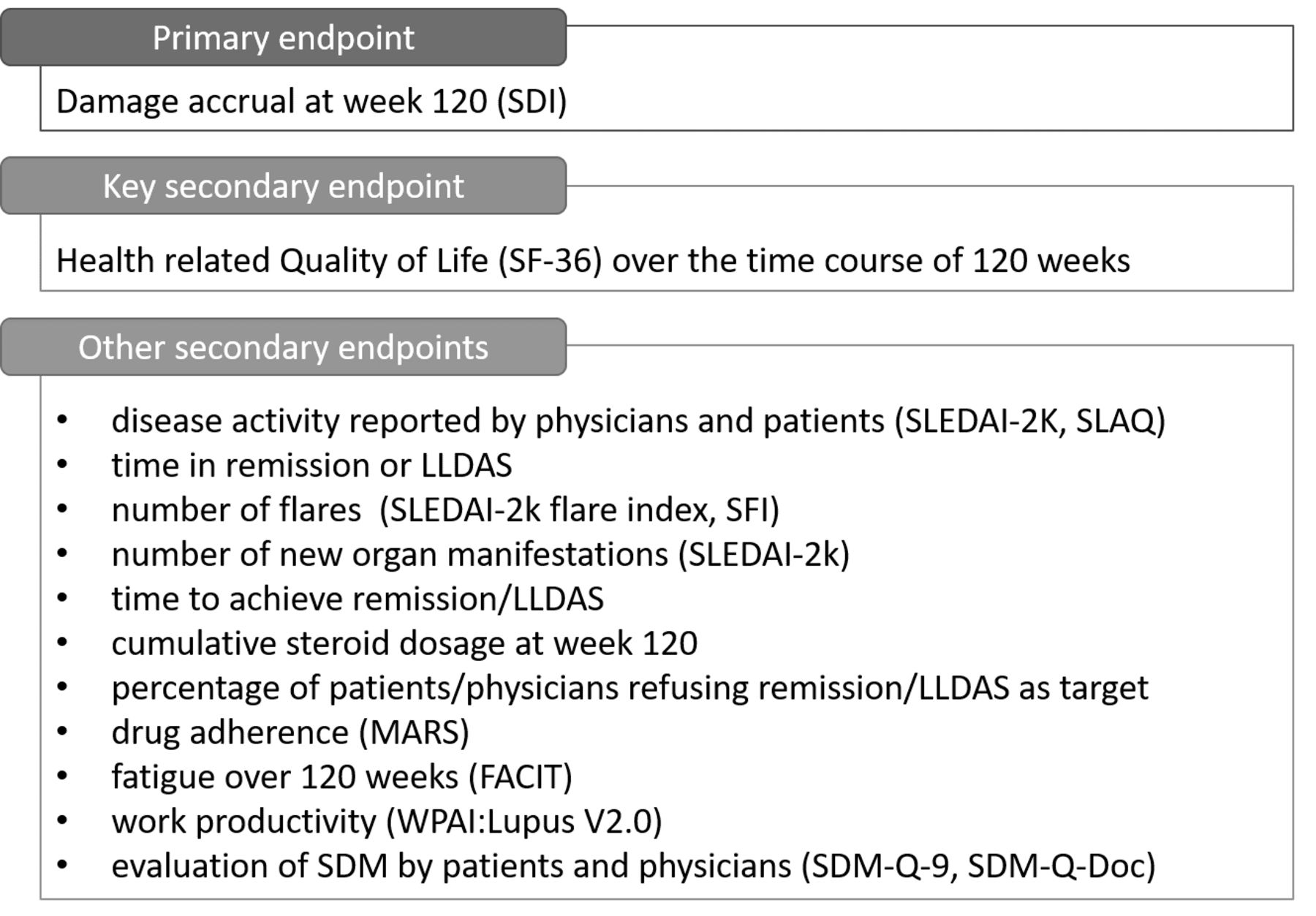
Primary and secondary endpoints (figure 2) will be evaluated at 120 weeks. The major outcome is the reduction in damage accrual (SLICC/ACR Damage Index, SDI). The patients’ relevant major secondary endpoint will be the HRQoL (36-Item Short Form Survey, SF-36).
{kind=link}
{kind=link}
Study endpoints. ACR, American College of Rheumatology; FACIT, Functional Assessment of Chronic Illness Therapy; LLDAS, Lupus Low Disease Activity State; MARS, Medication Adherence Report Scale; SDI, SLICC/ACR Damage Index; SDM, shared decision-making; SDM-Q-Doc, SDM Questionnaire-Physician's version; SDM-Q-9, 9-item SDM Questionnaire; SFI, SLEDAI Flare Index; SF-36, 36-Item Short Form Survey; SLAQ, SLE Activity Questionnaire; SLEDAI-2K, SLE Disease Activity Index 2000; SLICC, Systemic Lupus International Collaborating Clinics; WPAI, Work Productivity and Activity Impairment Questionnaire.
Justification of design aspects
Control/comparators
SoC was chosen as control as it reflects the clinical routine and allows for direct comparison of T2T versus routine treatment. The targets remission on therapy (ROT) and LLDAS will be used as comparators. They symbolise different goals for disease control and bear challenges that are important to evaluate (eg, GC reduction, amount of immunosuppression).
Dose, mode and scheme of intervention
We chose a 6-week interval over a period of 24 weeks for patients not in target to enable frequent clinical and laboratory check-ups in order to identify endangering situations and allow for short-term therapeutic adjustments as the foundation of T2T. Patients need to be stable in target for at least 6 weeks after a minimum of 24 weeks of tight control before changing to 12 weekly visits. A phase of stable disease is crucial to reduce the risk of sudden flares between subsequent 12 weekly follow-ups. In case of a flare, physician and patient decide to enter another tight control phase of 24 weeks. As this is the first trial on T2T in SLE, adaptation from T2T studies in RA was intended. However, follow-up intervals in these trials were 4 or 12 weeks and thus either too short or too long for the complex and potentially organ-threatening SLE. Thus, we chose the 6 weekly study visits and 12 weekly follow-up visits on recommendations and eminence-based experience. The period of 24 weeks tight control allows for small adjustments and was chosen to prevent overtreatment and a forced remission/LLDAS as the target does not have to be reached by the next visit. The study aim should be relevant to patients. Therefore, damage as surrogate of long-term outcome and HRQoL as a patient-reported outcome were chosen as endpoints. Damage accrues slowly and over time in SLE and follow-up-times need to be longer in damage-focused SLE trials, than in usual studies. Hence, a follow-up time of 120 weeks was chosen to allow for the detection of a clinically significant difference in damage accrual between the groups. SDM is crucial in T2T as patients should have the opportunity to decide about changes in treatment. SDM includes the option to reject T2T; some patients have LDA that can only be controlled by GC dosages slightly above the allowed threshold. However, in these cases, options to reach the target should still be explored at every visit.
The flexibility of the trial with the option to leave and re-enter tight control reflects clinical care, and presumably more patients will achieve the aimed target. Given that the T2T strategy is used regularly in clinical care of patients with SLE, the study is not limited to an inception cohort like most T2T trials in RA. In consequence, the individual medical treatment cannot be prescribed according to standard protocol. Still, pharmaceutical treatment decisions will be guided by a decision scheme based on the recently updated EULAR recommendations.17
Inclusion/exclusion criteria
The broadly defined inclusion and exclusion criteria allow for inclusion of the vast majority of patients with SLE treated in the participating centres and the study population will thus reflect and represent the heterogeneity of patients with SLE in real life. Patients of all genders and >18 years of age will be included; hence, generalisability is expected to the majority of adult patients with SLE.
Outcome measures
The prevention of damage accumulation and improvement of HRQoL by controlling disease activity and limiting the use of harmful drugs are our major aims. Best long-term outcome surrogate for SLE is damage (captured by the SDI, a validated, disease-specific index used to analyse the effects of ROT and LLDAS13 24). Thus, the primary trial outcome will be the SDI increase between (a) ROT and SoC and (b) LLDAS and SoC after 120 weeks. HRQoL is the key secondary outcome (measured by SF-36), as HRQoL is considered the most relevant parameter for patients. Other relevant factors listed above allow the evaluation of efficacy and feasibility of different steps and aspects of T2T in SLE. In addition, blood samples for biomarkers will be taken at study entry, after target achievement and the end of the study and stored in a biobank. To characterise the content of a patient’s visit in T2T versus control group, the patients will answer a questionnaire after every physician contact.
Methods against bias
The trial uses a cluster-randomised design, where the study centres constitute the single clusters. That is, all patients within a single study centre will be in the same treatment group and treated identically. We decided against individual randomisation on the patient level because this comes with a double risk of contaminating the intervention effect. First, patients within the same study centre will interact with each other and exchange information about the frequency and content of their treatments before and after their visits, and potentially also about the idea of T2T. Second, SDM and T2T training will influence the treating physicians in their approach and attitude towards the patients and their decisions, an effect that cannot deliberately be ‘switched off’ when treating patients who had been randomised to the control group. In contrast, actively deciding against T2T and reaching remission/LLDAS when treating a patient in the control group will be almost impossible for the treating physicians. Potential baseline imbalances of study centres will be reduced by matching them in strata of size 3 before randomisation using centres’ characteristics like patient numbers, hospital care type, type of services, existence of a multidisciplinary team and regional characteristics thus maximising homogeneity of centres within a matching stratum while maximising heterogeneity across matching strata. Randomising centres to one of the treatments will then be performed within each matching stratum. Blinding of patients for their aim is impossible, as SDM is part of T2T. Nonetheless, provided information regarding trial design and targets will be standardised and kept to a minimum. To secure similarity across all intervention centres, standardised training is mandatory for all team members. Disease heterogeneity will be addressed by including a high number of patients with potentially every disease manifestation to obtain a representative study cohort. A data monitoring committee will regularly monitor recruitment and application of tight control and T2T and will communicate irregularities.
Sample size calculation
Sample size calculation was performed with respect to the primary endpoint, damage accrual as measured by the SDI, and was based on a two-tailed t-test (α=0.05) with Bonferroni adjustment for two comparisons (LLDAS vs SoC and remission vs SoC) and power of 80% (ß=0.2). We assumed an SD of the SDI of 0.89,25 and a minimal clinically relevant difference in SDI of 0.3 points. This reduction in SDI corresponds, for example, to prevent one out of three patients from damage accrual or to avoid failure or insufficiency of one organ out of three affected organs. The design effect (Deff) for cluster-randomised controlled studies was taken into account as described by Murray.26 For calculating Deff, an average cluster size of 70 patients and an intracluster correlation of 0.015 were assumed.27 Based on these assumptions, a sample size of 1060 study participants was calculated. To allow some moderate dropout (20%), 1272 participants split up into three treatment arms (ie, 424 patients per arm) will be included into the study.
Statistical analysis
The statistical analysis for the primary outcome will be accomplished within a hierarchical (mixed) model. This will include the SDI at week 120 as the outcome, the intervention as well as baseline SDI, age, disease duration and gender as covariates, and two random intercept effects (one for the matching/randomisation stratum and for the single study centre) to account for the hierarchical/clustered structure of data.26 Corresponding to the sample size calculation, the two primary comparisons (LLDAS vs SoC and remission vs SoC) will be confirmatorily tested to the 2.5% level to ensure overall type I control to the 5% level. Analyses will be performed following the intention-to-treat principle. Missing values will be accounted for by multiple imputation following the ideas and guideline of Sterne et al.28 There will be no planned interim analysis for efficacy and no confirmatory subgroup analyses. The secondary outcomes will be analysed analogous to the primary outcome by the corresponding hierarchical model.
Moreover, the study data will be analysed in an explanatory way by using different data science and machine learning-based approaches to reveal additional insights into the collected data and thereby to obtain potential vital design aspects, which could guide the future treatment of patients with SLE. The idea is to use machine learning especially in a hypothesis-free way to enable to identify new and relevant patterns and to formulate additional data-driven hypotheses for further studies. One important task will be to obtain subgroups of patients within each arm via the application of different supervised or unsupervised clustering and classification approaches. Different subgroups might have distinct characteristic profiles, for example, in relation to disease activity and patient-reported outcomes. Another research task will be to investigate via machine learning-based approaches whether the length of remission/LLDAS phases of a patient can be predicted based on the data being collected at different time points before the event occurs. This might deliver first hints in relation to visit frequencies for potential future studies.
Ethics and dissemination
The study will be performed in accordance with the Declaration of Helsinki and Good Clinical Practice. Approval will be obtained from the ethical committee of all participating centres separately. Informed consent will be obtained from each patient. Data will be entered pseudonymously, and the decryption key will be stored separately from the data. Data will be stored for 15 years after closing of the databank. All patients will receive standard SLE treatment and no experimental drugs will be administered, so that the study-related risk is minimal. However, the frequent visits and treating to target might bear the risk of study-related intensified treatment in that immunomodulatory treatments are initiated in stages in which the SoC approach would probably consist of ‘watchful waiting’. Earlier initiation of treatment and frequent visits could on one hand lead to early detection and control of subclinical disease activity. Reaching the target early is expected to result in low disease-related damage and better HRQoL. On the other hand, intensified treatment increases the chance of drug-related adverse events and a patient might receive treatments for manifestations that could have remitted spontaneously due to natural course of the disease. However, the six weekly study visits allow for close monitoring of adverse events, thus drug-related toxicity should be detected early. In patients with fast remission of manifestations that might not have required therapy, treatment can be reduced quickly due to the frequent visits. Intensified treatment in the intervention group may include higher steroid dosages, too. To prevent high-cumulative steroid dosages, a steroid reduction plan for the following 6 weeks has to be recorded and handed out to the patient. Another risk is a fast reduction of steroids to reach the target. For this reason, 24-week loops were inserted to provide time to slowly approach the target and to not feel the need to abruptly reduce or stop steroid treatment. However, lower doses of steroids are part of the target and are associated with less side effects and reduced damage. Each participant will have the right to discontinue the study at any time and to request the deletion or anonymisation of his/her data. A study-specific insurance covering the travels will be provided to all participants.
The patients’ safety will be assessed and documented at each study and follow-up visit. Risk-adapted safety surveillance of this clinical study will be conducted by the independent Coordination Center for Clinical Trials of the Faculty of Medicine at the Heinrich-Heine-University Düsseldorf (KKS Düsseldorf). This includes management and reporting of study treatment-related adverse events and regular safety reporting by the KKS Düsseldorf for assessment by an independent data safety monitoring board and for the respective ethics committees.
Data will be archived according to standards of drug trials for 15 years. Individual participant data that underlie published results (text, table, figures and appendices) will be made available after deidentification together with the study protocol, statistical analysis plan and the analytics code beginning 3 months and ending 5 years following the results’ publications to researchers who provide a methodologically sound proposal in order to achieve the proposal’s aims. Data will be available for 5 years at a third-party website.
Discussion
As uncontrolled disease activity and GC use (>7.5 mg/day prednisolone) are the major causes of damage and death in SLE,29 30 their consistent control by T2T could prevent these fatal outcomes. T2T provides a protocol both physicians and patients can use as guidance, which will be led by SDM. Inability to reach the target will also reveal patients with the specific need for additional interventions. The first aim of implementing T2T in SLE is to reduce patients’ burden of illness by reducing disease-related and treatment-related long-term damage and improving HRQoL. Futile treatments will be shortened due to frequent reassessments and therapeutic adjustments. Comparison of remission and LLDAS will help to identify the target with the best benefit–risk ratio concerning attainability, adverse events and damage. The emphasis on SDM will strengthen patient autonomy and will improve both their satisfaction and medical condition.
Direct costs might rise due to an increased application of immunomodulatory drugs including biologics. On a long-term basis, these costs should be balanced out with reduced costs due to less organ damage and improved participation and adherence. Indirect costs are expected to be reduced by less dependency on physical and mental support and a lower rate of work disability resulting in early retirement. It is intended to transfer the highly standardised and validated data collected during the trial into a national registry for patients with SLE to facilitate further trials and investigations, thus improving healthcare and management of patients with SLE.
Assuming the trial will confirm the benefit of T2T in SLE, the impact on clinical practice will be fundamental for both national and international SLE centres as well as private practices. The results will also be transferred to new (inter)national guidelines and recommendations. As LUPUS-BEST will also identify patients who do not response to the approved medications, the requirement profile for new interventions can be defined. This will stimulate new trials and trial designs following the precision medicine approach.
The expected benefit for the individual patient is a reduction in damage accrual, improved HRQoL and higher life expectancy. If the trial is successful, the T2T concept will be transferred to patients outside the trial. In addition, SDM promotes the patients’ autonomy and patients might benefit over time from a more profound knowledge about treatment strategies and the efficacy of drugs in specific situations.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
Ethics statements
Acknowledgments
We thank the participating centres: Charité Universitätsmedizin Berlin, Rheumazentrum Ruhrgebiet Herne, Ruhr University Bochum, University Medical Centre TU Dresden, University Clinic Erlangen, Kliniken Essen Mitte, University Clinic Frankfurt, University Clinic Freiburg, Justus Liebig University, Campus Kerckhoff, Bad Nauheim, Medical University Hannover, University Clinic Heidelberg, Centre for Psychosocial Medicine Heidelberg University, UKSH Campus Kiel, University Clinic Mainz, LMU Munich, University Clinic Würzburg, Fraunhofer Institute for Intelligent Analysis and Information Systems (IAIS) and the Lupus Erythematodes-Selbsthilfegemeinschaft e.V. for their careful contribution and revision of the protocol.
References
Footnotes
Twitter @JoMucke
Contributors JM and MS contributed equally to the trial planning and drafting of the manuscript. OK and RB did the sample size calculations and statistical planning of the trial. All authors carefully revised the manuscript and agreed on the final version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests JM received consulting and lecture fees from AbbVie, Amgen, AstraZeneca, BMS, Celgene, Gilead Sciences, Galapagos, GlaxoSmithKline, Janssen-Cilag, Lilly, Medac, Mylan and Novartis Pharma. MS received consulting and lecture fees from MSD, Abbott, AbbVie, Pfizer, GlaxoSmithKline, UCB, Roche, AstraZeneca, Lilly, Janssen-Cilag, Sanofi-Aventis, Chugai, Celgene, Novartis, Boehringer Ingelheim and Bristol-Myers Squibb. RB, OK and SS declare no competing interests.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.