Article Text

Abstract
Objective To examine the long-term changes in circulating B cell subsets and IgG levels at 5+ years of continuous belimumab treatment and their correlations with efficacy and safety measures.
Methods This was a post hoc analysis of a continuation study (BEL112233; NCT00724867) of eligible US patients who completed the 76-week BLISS-76 Study (BEL110751; NCT00410384), with up to eight calendar-years of follow-up and median (IQR) belimumab exposure of 310 (209, 364) weeks. From week 76, patients initially randomised to intravenous belimumab 1 mg/kg or 10 mg/kg every 4 weeks in BLISS-76 continued to receive the same dose in the continuation study, while those initially randomised to placebo received belimumab 10 mg/kg intravenous every 4 weeks during continuation. All patients continued to receive standard SLE therapy. Biomarker data were collected, and the effects on baseline and early changes (weeks 0–24 after starting belimumab) from baseline in biomarkers on SLE Responder Index (SRI-4) and infection rate were evaluated.
Results Of the 819 patients from BLISS-76, 268 self-selecting patients entered BEL112233. Compared with baseline, B cell subset counts decreased by 40%–99% after 312 weeks (6 years), and serum IgG levels decreased by 28% after 284 weeks. Higher baseline naïve B cell counts were associated with greater SRI-4 response rates (p<0.05), whereas higher baseline SLE subset plasma and short-lived plasma B cell counts were associated with lower SRI-4 response rates (p<0.05). Elevated baseline IgG levels were associated with increased infection rates over the treatment period (p<0.05), and early greater decreases in IgG levels were associated with higher SRI-4 response rates (p<0.05).
Conclusions Belimumab treatment up to 312 weeks (6 years) resulted in substantial decreases in several circulating B cell subsets and IgG levels. Higher baseline naïve B cell counts and IgG levels were associated with improved SRI-4 response and increased infection rates, respectively.
- autoimmune diseases
- B-lymphocytes
- biological products
- lupus erythematosus
- systemic
Data availability statement
Data are available upon reasonable request. Anonymised individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
B lymphocyte stimulator (BLyS) is essential for B cell maturation and Ig class switching and production.
Overexpression of circulating BLyS correlates with worsening SLE disease activity and increased risk of SLE flares.
Belimumab (anti-BLyS) therapy has been evaluated for long-term efficacy and safety and was shown to reduce Ig levels and several circulating B cell subsets in the BLISS-76 Trial.
What does this study add?
In this post hoc continuation analysis of the BLISS-76 Trial, belimumab treatment up to 312 weeks (6 years) led to decreases in B cell subsets and IgG levels. Elevated baseline naïve B cells correlated with improvements in SLE Responder Index response, while elevated baseline IgG titre was associated with poorer SLE Responder Index outcome and greater infection rates.
How might this impact on clinical practice or future developments?
Assessment of B cell and IgG levels before initiating long-term belimumab therapy may facilitate prediction of response and infection risk of patients with SLE.
Introduction
SLE, a chronic autoimmune disease that affects multiple organs, is driven by the abnormal activation of B and T lymphocytes, and is associated with elevated titres of autoantibodies.1 2 B lymphocyte stimulator (BLyS; also known as B cell activating factor, or BAFF) is a ligand of the tumour necrosis factor cytokine family that is expressed as membrane-bound or soluble protein. BLyS is a vital B cell survival factor with important roles in B cell maturation and Ig class switching and production.3–5 Overexpression of BLyS is thought to be linked to the pathogenesis of SLE and other autoimmune diseases,6 and increased concentrations of circulating BLyS are correlated with worsening SLE disease activity that is indicative of mild-to-moderate flares. Furthermore, elevated (≥2 ng/mL) levels of BLyS were found to be predictive of SLE flares in a post hoc analysis of BLISS-52 and BLISS-76 belimumab trials.7
Belimumab is a human IgG1κ monoclonal antibody that is approved in the USA, the European Union, and Japan for the treatment of patients ≥5 years of age with active, autoantibody-positive SLE receiving standard therapy.8–10 Recently, belimumab was approved by the FDA for the treatment of adults with active lupus nephritis receiving standard therapy.8 Belimumab inhibits the activity of soluble BLyS and may restore the ability of autoimmune B cells to undergo apoptosis in a partially selective manner, ultimately leading to reduced SLE disease activity.1 11 Previous studies of up to 76 weeks (1.5 years) of belimumab therapy have demonstrated a sustained reduction in Ig levels and in several circulating B cell subsets, including naïve and activated B cells. Concurrently, reductions in SLE Responder Index (SRI-4), including Safety of Estrogens in Lupus Erythematosus National Assessment version of the SLE Disease Activity Index (SELENA-SLEDAI), newly occurring British Isles Lupus Assessment Group A and B organ domain scores, Physician’s Global Assessment, and incidence rate of severe flares were observed.12–15
A 7-year assessment of patients who completed the BLISS-76 Trial (GSK study BEL110751; ClinicalTrials.gov identifier: NCT00410384) demonstrated the long-term efficacy and safety of belimumab.13 Longer-term reductions in autoreactive B cells and Ig titres may facilitate achieving the treatment goals of SLE therapy, which include reducing disease activity, preventing flares and long-term organ damage, and reducing corticosteroid exposure. As such, further investigation of the long-term impact of belimumab on biological markers of SLE, and how these may correlate with measures of disease activity and organ damage, is of considerable clinical interest15 but is yet to be fully explored. Therefore, the purpose of the present analysis was to examine long-term changes in biomarkers, including B cell/B cell subset counts and IgG titres, for up to 312 weeks (6 years) of continued belimumab treatment.
Methods
Study design
This was a post hoc analysis of a multicentre, continuation study (GSK study BEL112233; ClinicalTrials.gov identifier: NCT00724867) of eligible patients in the USA who completed the 76-week phase III BLISS-76 parent study,12 conducted from 5 August 2008 to 26 March 2015. In BLISS-76, patients received intravenous belimumab 1 mg/kg or 10 mg/kg, or placebo, plus standard therapy (eg, corticosteroids, immunosuppressants and/or antimalarials), every 4 weeks for 72 weeks, with a final evaluation at week 76. Patients who completed the BLISS-76 Study had the option to enrol in the continuation study, where patients who had received belimumab remained on the same dose, while those who had previously received placebo were switched to belimumab 10 mg/kg intravenous. Of note, for patients who had received placebo in BLISS-76, day 1 was the day of their first dose of belimumab. Following a protocol amendment on 9 March 2011, patients receiving belimumab 1 mg/kg had their dose increased to 10 mg/kg. All belimumab doses were pooled for this analysis.
The study end date was five calendar-years from the date of enrolment of the last patient into the continuation study. Clinical site personnel were blinded to the BLISS-76 parent study treatment until results of the parent study were made public. Written informed consent was obtained from all patients prior to enrolment.
Patients
Patients were assessed for eligibility to enrol in the continuation study at the week 72 visit of BLISS-76 and were required to receive the first continuation-study belimumab dose on day 0 of the BEL112233 continuation study and within 2–8 weeks after the last dose of investigational product in BLISS-76. Excluded patients included those with clinical evidence of significant, unstable or uncontrolled acute or chronic disease not due to SLE (in the investigator’s opinion), those who experienced in BLISS-76 any adverse event (AE) that could put the patient at undue risk (in the investigator’s opinion), or those who did not meet the enrolment criteria.
Biomarkers assessment and correlative analyses with SLE disease activity and safety
Assessed biomarkers included change from baseline in circulating concentrations of B cells (CD19+ and CD20+), naïve B cells (CD19+/CD20+/CD27−), memory B cells (CD19+/CD20+/CD27+), activated B cells (CD20+/CD69+), plasmacytoid B cells (CD19+/CD20+/CD138+), SLE subset plasma cells (CD19+/CD27bright+/CD38bright+),16 short-lived plasma B cells (CD19+/CD20−/CD27bright+), and plasma B cells (CD19+/CD20−/CD138+) over 312 weeks (6 years), and IgG over 284 weeks (~5.5 years). All samples were analysed, and subsequent B cell subset quantification was performed at Q2 Solutions Laboratories as described in the online supplemental methods.
Supplemental material
The discrepancy between B cell and IgG level data availabilities derives from differences in the frequency of the measurement of the two biomarkers: while all B cell subsets were measured at baseline and every 24 weeks after first belimumab dose in the continuation study, IgG levels were measured only every 48 weeks (except for study year 1 when measurements were also taken at the week-24 visit) plus at an additional 8-week follow-up visit after the last dose. The 312-week versus 284-week difference derives from patients exiting the study mid-study year without a follow-up visit.
The effects of baseline and early changes (weeks 0–24 of the continuation study) from baseline in biomarkers on SRI-4 responses17 and the overall number of serious or severe infections (ie, AEs of special interest in the context of chronic B cell suppression) were evaluated.
Statistical analyses
Analyses were performed on the modified intention-to-treat (mITT) populations from BLISS-76 and its long-term continuation study BEL112233, defined as patients who had received belimumab 1 mg/kg or 10 mg/kg intravenous or placebo. For patients receiving belimumab in BLISS-76, data on biomarker, disease activity and safety from baseline to week 76 were included. For those receiving placebo in BLISS-76, only data collected in the continuation study (after they had switched to belimumab) were included. Thus, for patients who had received placebo in BLISS-76, day 1 was the day of their first belimumab dose. Continuous and categorical data were summarised using means and SD, and counts and percentages, respectively. Tertiles of absolute B cell count and IgG levels as well as tertiles of change from baseline in these measurements were calculated at the end of each 24-week interval in the continuation study. To understand the univariate relationships between tertiles of B cells and IgG levels and efficacy end points, Spearman correlation coefficients and their 95% CIs were calculated. Statistical analyses were conducted for each B cell subset and IgG. The main comparisons were tertile 1 versus tertile 2 and tertile 1 versus tertile 3 for the efficacy end point (ie, SRI-4 response and the occurrence of serious or severe infections).
The rates of SRI-4 response and severe or serious infections were analysed via separate negative binomial regression models for each biomarker with offset log of follow-up time and terms for age, body mass index (BMI), baseline SLEDAI, baseline immunosuppressant use, treatment group, and either baseline biomarker tertile or change from baseline biomarker tertile. Incidence rate ratios and their associated 95% CIs were estimated from these models for each biomarker and covariate of interest, and similar analyses were conducted to determine whether early changes (weeks 0–24 after starting belimumab) from baseline in biomarkers were predictive of SRI-4 response and severe or serious infections.
Patient and public involvement
We did not involve patients and/or the public in the design of this study.
Results
Patients
The global mITT population comprised 819 patients with SLE from the parent BLISS-76 Study (BEL110751), of whom 268 self-selecting patients (from the US sites only; n=177 receiving belimumab and n=91 receiving placebo in the parent study) of the 576 patients who completed BLISS-76 subsequently entered the continuation study (BEL112233). Patient disposition and baseline demographics of the BLISS-76 population who entered the continuation study are shown in table 1.
Baseline characteristics of patients in the continuation study (n=268)
Among those who entered the continuation study, 93.3% (n=250/268) were female and mean (SD) age was 42.8 (11.3) years. In total, 69.4%, 21.3% and 9.3% of patients were White, of Black African ancestry/African American or other race, respectively. Overall, patient characteristics were similar between the parent and continuation study populations, and baseline characteristics between placebo and belimumab treatment groups in the parent study were similar. Over the period of both studies, up to eight calendar-years of data were collected, with a median (IQR) belimumab exposure of 310 (209, 364) weeks.
Biomarker responses over time
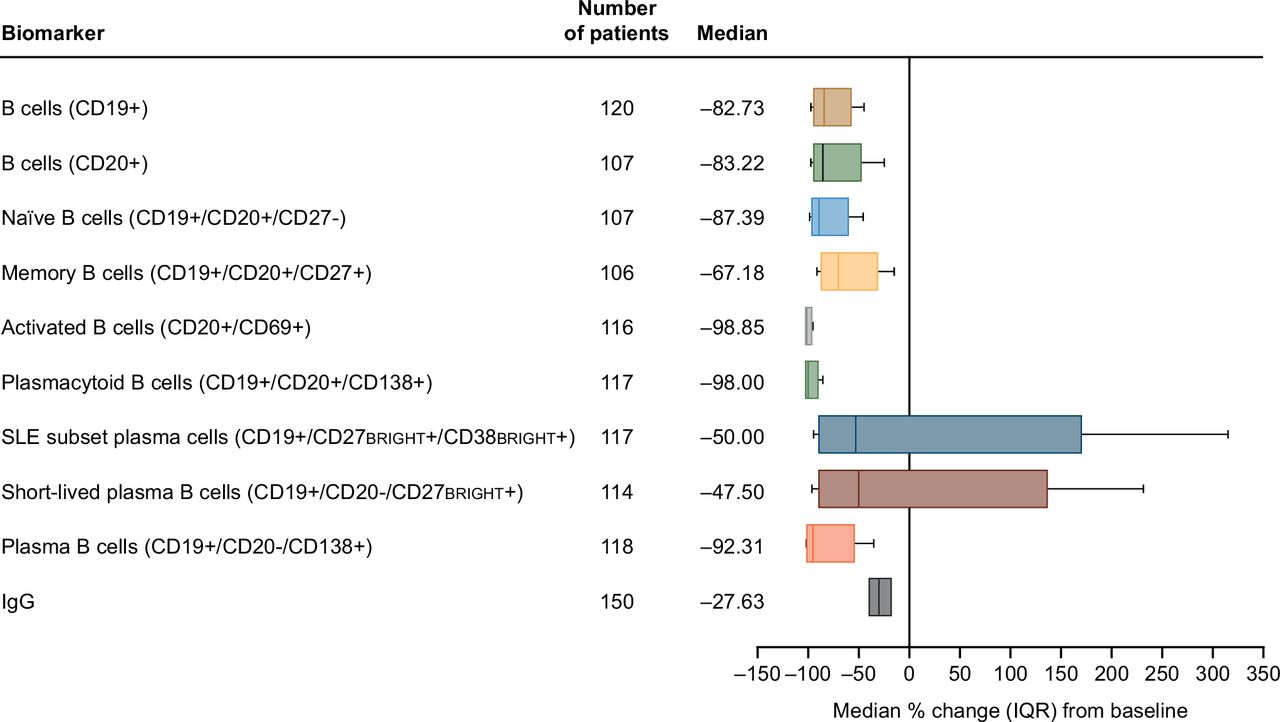
After 312 weeks (6 years) of continued belimumab dosing (study year 7, week 24), net reductions from baseline of 83%–92% in circulating CD19+, CD20+, and naïve and plasma B cells, 98%–99% in activated and plasmacytoid B cells, 67% in memory B cells, and 40%–50% in the SLE subset plasma and short-lived plasma B cells were observed (figure 1). The wider range in SLE subset and short-lived plasma B cell reduction compared with other types of B cells was likely due to the comparatively small base numbers of these subsets in the overall B cell population, leading to a higher variability in count. Following 284 weeks (~5.5 years) of belimumab (study year 6, week 48), a 28% reduction from baseline in serum IgG levels was observed (figure 1). Circulating concentrations of all B cell types, except memory B cells, decreased rapidly from baseline in the first 24 weeks of belimumab treatment and then declined more slowly through to week 72 (figure 2). In contrast, memory B cell counts markedly increased during the first 24 weeks, then subsequently declined progressively over time (figure 2).
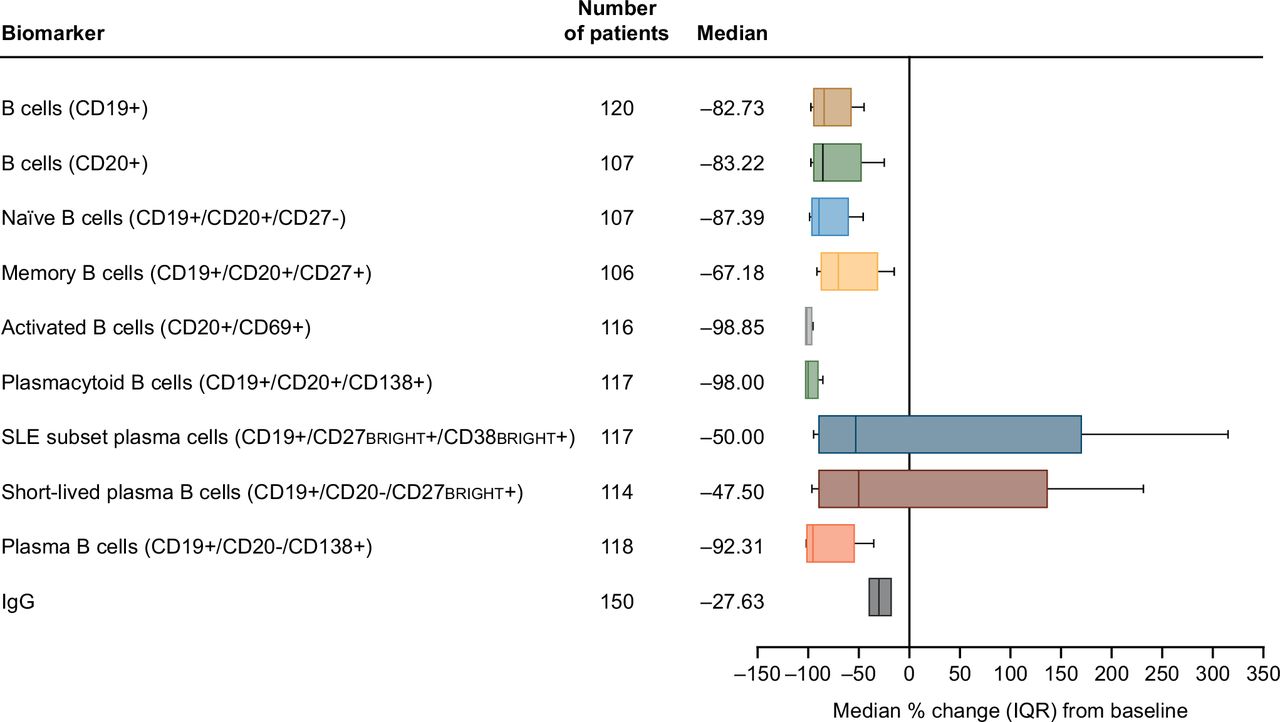
Median percentage change (IQR)* from baseline at last time point† for B cell counts and serum IgG levels‡ in patients treated with belimumab (mITT population). *Whiskers indicate 10th and 90th percentiles; †study year 7, week 24 for all B cell subsets, and study year 6, week 48 for IgG; ‡10th and 90th percentiles for the IgG data are not shown. CD, cluster of differentiation; IQR (25th and 75th percentiles); mITT, modified intention-to-treat.

Median percentage change from baseline over time for B cells and IgG in patients treated with belimumab (mITT population). CD, cluster of differentiation; mITT, modified intention-to-treat.
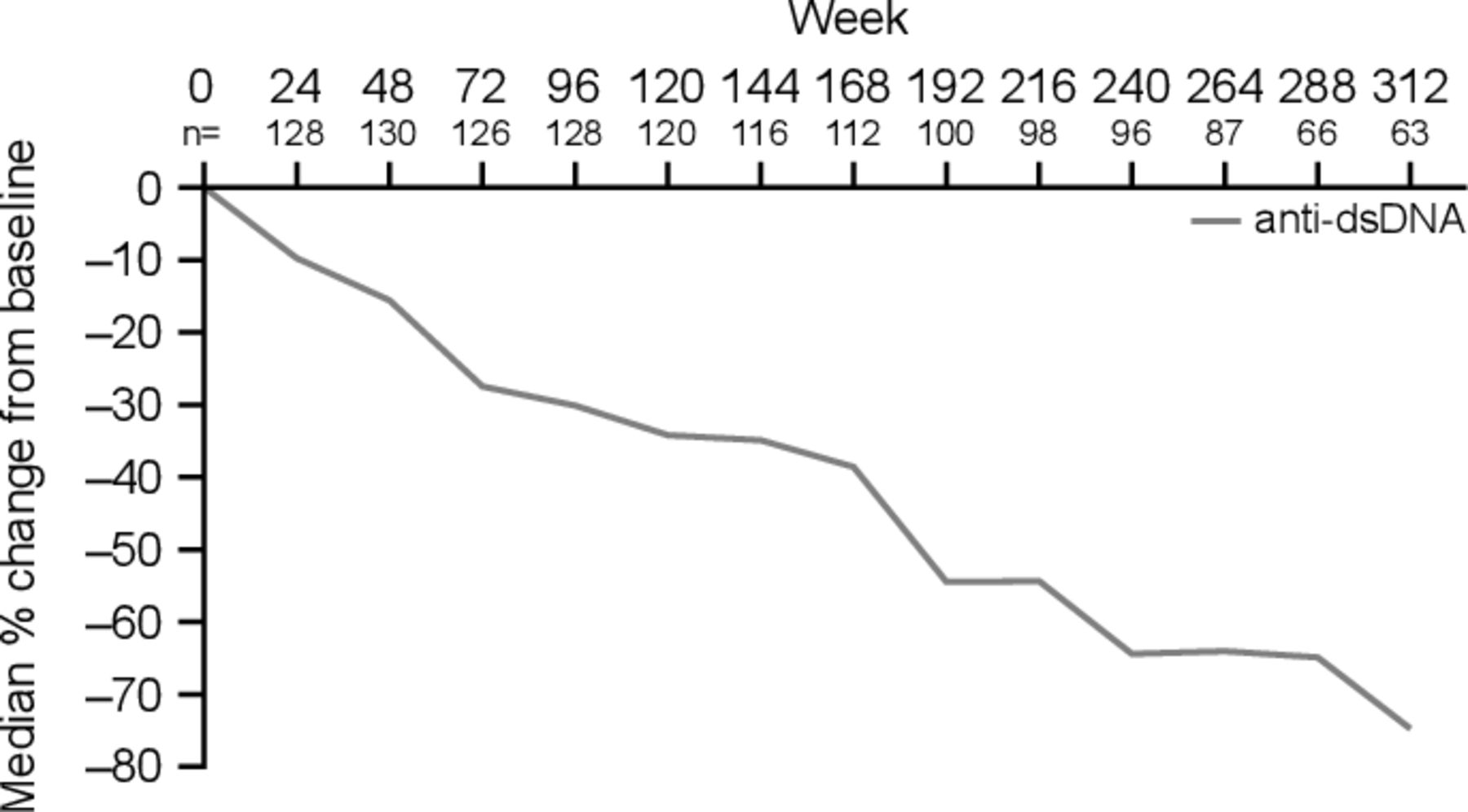
Beyond 76 weeks (1.5 years) of treatment, B cell numbers either continued to gradually decrease (CD19+, CD20+, memory, activated, plasmacytoid, and plasma B cells) or stabilised (naïve, SLE subset and short-lived plasma B cells; figure 2). By weeks 288 or 312, the decline in numbers of most B cell subsets (naïve, activated, plasmacytoid and plasma B cells) had plateaued, while memory B cells continued to demonstrate a sustained decline, although at a slower rate than at earlier time points. Serum IgG levels continued to progressively decrease throughout the entire study period (figure 2). Anti-double-stranded DNA (anti-dsDNA) IgG antibody levels in a subpopulation of patients (n=135) positive for anti-dsDNA antibodies (≥30 IU/mL) at baseline also decreased progressively over the follow-up period, but with larger reductions relative to baseline than were observed for total IgG levels (figure 3).
{kind=link}
{kind=link}
{kind=link}
Median percentage change from baseline over time for anti-dsDNA in patients positive for anti-dsDNA at baseline (≥30 IU/mL) treated with belimumab (mITT population)* *The anti-dsDNA data presented are from the clinical study report of study BEL112233 (NCT00724867) for a subpopulation of patients (n=135) positive for anti-dsDNA antibodies (≥30 IU/mL) at baseline dsDNA, double-stranded DNA; mITT, modified intention-to-treat.
Baseline biomarker correlation to clinical response or infection risk
Biomarker changes during belimumab treatment were explored for associations with improvement in SRI-4 and with an increased risk of infections.
SRI-4 response
Higher naïve B cell counts at baseline were associated with greater SRI-4 response rates to belimumab (rate ratio per tertile increase 1.10; 95% CI 1.01 to 1.21, p<0.05), whereas higher baseline SLE subset plasma and short-lived plasma B cell counts were associated with lower SRI-4 response rates to belimumab (rate ratio per tertile increase 0.86; 95% CI 0.79 to 0.95 and 0.88; 95% CI 0.80 to 0.96, respectively; each p<0.05) (table 2). Other assessed B cell subsets or IgG at baseline did not significantly correlate with changes in SRI-4 response rates (table 2).
Impact of baseline B cells and IgG on efficacy and safety over the entire treatment period (mITT population)
Supplemental material
Serious or severe infections
Overall rates of serious or severe infection were low and stable across the entire study period (five events occurred, corresponding to a rate of serious or severe infection for any time postbaseline of 0.3 per 100 patient-years), and ranged from 0.4 per 100 patient-years at study years 0–1 to 0 per 100 patient-years at study years 6–7. In total, there was one case each of bacterial sepsis (occurring in study years 4–5; rate of <0.1 per 100 patient-years), genital herpes (occurring in study years 0–1; rate of <0.1 per 100 patient-years), herpes zoster (occurring in study years 1–2; rate of <0.1 per 100 patient-years), and 2 cases of septic shock (occurring in study years 2–3 and 5–6, for a combined rate of 0.1 per 100 patient-years). Elevated serum IgG levels, but not B cell counts, at baseline were associated with increased rates of infection over the treatment period (rate ratio per tertile increase 1.75; 95% CI 1.24 to 2.46, p<0.05) (table 2).
Impact of early biomarker changes
Changes in SRI-4 response or infection rates did not appear to be associated with early (within the first 24 weeks of belimumab treatment) changes in biomarkers, except for serum IgG levels. Patients with small changes in serum IgG in the first 24 weeks of belimumab treatment also showed low SRI-4 response rates (rate ratio per tertile increase 0.90; 95% CI 0.82 to 0.98, p<0.05) (table 3).
Impact of early changes in B cells on efficacy and safety over the entire treatment period (mITT population)
Impact of covariates on end point-biomarker relationship
Baseline SELENA-SLEDAI Scores ≥10 were associated with a significant increase in SRI-4 responses compared with lower scores (<10), with a rate ratio estimate of ~1.5 in all statistical models. Age and BMI, and the use of immunosuppressants, did not significantly predict SRI-4 responses. Similarly, baseline SELENA-SLEDAI category, immunosuppressant use and BMI did not have a significant impact on the incidence of serious or severe infections over the treatment period. Age was not a significant factor in any of the B cell statistical models but significantly impacted the number of serious or severe infections in the IgG statistical models (rate ratio: 1.37; 95% CI 1.05 to 1.78, per 10 year increase in age, p=0.020).
Discussion
The present analysis builds on previous studies characterising biomarker responses over 2 years and beyond, and clinical responses over 6 years with belimumab therapy.12–15 We observed a marked decline in circulating B cell subset counts during such long-term, continual belimumab treatment, but no subsets were completely depleted. This is in line with observations from BLISS-7612 and phase II studies18 19 of belimumab. B cell subsets show varying degrees of dependence on BLyS, so it is anticipated that their response to belimumab treatment will differ accordingly. In the present study, most B cell subset counts reached a plateau by weeks 288 or 312, and with the exception of memory B cells and serum IgG levels, no further substantial reductions would be expected. Of note, neither SLE subset nor short-lived plasma B cell counts plateaued and instead fluctuated throughout the study period. This, however, may be due to the higher variability in blood cell count for these rare/small subsets.
An initial increase in memory B cells in response to belimumab, peaking at week 24, was followed by a sustained decline, and a similar pattern has generally been observed in earlier studies of belimumab.13 15 18 The cause of this response is unclear; the time profile is consistent with a transient redistribution of memory B cells from lymphoid tissues to the circulation, combined with a slow decrease in total numbers. The results presented (ie, a substantial reduction of memory B cells after multiple years of treatment) are in contrast with reports of their limited dependence on BLyS for survival.20 21 However, this seeming discrepancy could be explained if the survival of memory B cells is BLyS-independent but the survival of their precursors is not. In that case, after BLyS reduction by belimumab, the pool of memory B cells would not be adequately replenished and would decrease over time.
Low IgG levels are considered a risk factor for infections.22 In this study, a median reduction of IgG levels of ˂30% to a median level of 10.30 g/L (8.57 g/L at 25th percentile) after approximately 5.5 years of belimumab treatment did not raise safety concerns for a typical patient, based on IgG lower limits of normal of 6─7 g/L, a result in line with previous reports.23 During the same time frame, anti-dsDNA IgG antibody levels in patients positive for anti-dsDNA antibodies at baseline decreased by 64.9% (figure 3), demonstrating that long-term belimumab treatment preferentially decreased these SLE-specific autoreactive antibodies.
B cell reductions from baseline were not correlated with any significant changes in the known safety profile of belimumab. Despite continued reductions in B cell subsets, there was no trend for increased rates of serious or severe infections with longer treatment durations.23 24 This suggests that the residual B cell populations retained B cell clones essential for humoral immunity and that belimumab might preferentially reduce B cells with antigen specificities or functionality redundant for humoral immunity.25 However, in the present analysis, elevated serum IgG levels at baseline were associated with significantly increased rates of infection over the treatment period. This may be related to patients having more severe disease (ie, high baseline SELENA-SLEDAI Scores) at study entry. Higher disease activity and more refractory disease may in turn increase the likelihood of patients receiving more potent and higher doses of immunosuppressive therapies and, therefore, having an increased susceptibility to infections. While this hypothesis could not be confirmed within the confines of this study, such a relationship of serological activity (low complement levels and elevated titres of anti-dsDNA antibodies) with increased baseline IgG, SELENA-SLEDAI Scores, and immunosuppressant use was demonstrated previously in the pooled BLISS-52 and BLISS-76 population.26 Long-term extension studies showed reductions in immunosuppressant use with belimumab, and lower incidence of infections, including serious infections.23 24 In the current study, immunosuppressant use did not have a significant impact on rates of infections.
Smaller reductions or increases in serum IgG levels in the first 24 weeks were associated with significantly lower SRI-4 response rates to belimumab. This finding is consistent with an analysis of biomarkers over the first 76 weeks in which belimumab-treated patients with normalisation of IgG levels had significantly greater SRI response rates than did patients without normalisation at weeks 24 and 40.15
In the present study, elevated baseline naïve B cell counts were associated with improved SRI-4 response rates, while higher baseline SLE subset plasma and short-lived plasma B cell counts were associated with poorer SRI-4 response rates. None of the other B cell subsets significantly influenced SRI-4 response. Improvements in clinical response could be due in part to reductions in certain BLyS-dependent B cell populations leading to a reduction in inflammatory cytokines27 and/or autoantibody production.16 18
Higher baseline SELENA-SLEDAI Scores (≥10 vs 6–9) used as covariates on end point-biomarker relationships were associated with a higher SRI-4 response rate. This relationship has been shown elsewhere in univariate analyses of likelihood of response and baseline disease activity and may reflect the effect of analysing an absolute threshold (4-point reduction as part of the SRI-4) with more room for improvement or a greater involvement of BLyS-driven pathology in patients with high-disease activity as compared with patients with lower-disease activity.26
There are several limitations inherent to the study. As a post hoc analysis, the study was not formally designed to assess the relationship between numbers of B cells, IgG concentrations, and efficacy and safety end points. However, assessment of absolute numbers/levels of these biomarkers would be of interest to evaluate in the future, because this may allow potential thresholds to be established to assist clinicians in relating biomarker levels to infectious events or other AEs observed in clinical practice. At the time of the study, the differentiation between naïve B cells and double-negative memory B cells had not been well established and, accordingly, was not studied. As such, further investigation is warranted. However, a recent analysis of samples from patients with lupus nephritis or with SLE demonstrated that both unswitched and switched (IgG1+, IgG2+, IgA1+, IgA2+) memory B cells increased after belimumab treatment.28 Increases of both preswitched (IgD +CD27+) and postswitched (IgD−CD27+) memory B cells have also been demonstrated for another BLyS/BAFF antagonist.29 At baseline, there may have been an existing effect of prior therapies on B cell subsets, and no adjustments for multiplicity were made, increasing the chance of introducing a type 1 error. When analysing the B cell counts and serum IgG levels after 312 weeks (6 years) of belimumab treatment, the completers subpopulation represents <45% of the 268 patients who initially entered the continuation study. This subpopulation may not be representative of the entire study population, as there may have been a natural selection bias towards those who had good initial responses to belimumab and few AEs, including infection. This may have contributed to the low rates of serious or severe infection as an AE. As such, these data should be interpreted with caution. However, this study’s design is similar to that of the two belimumab phase II continuation studies, in which patients also switched treatment (from placebo to belimumab or from lower to higher belimumab dose). In line with results from this study, the two phase II studies showed increases in SRI responses, and reductions in flares, AE rates and serological biomarker levels, over time.23 24 This analysis only focused on the B cell population in the blood and did not analyse B cell subsets in the secondary lymphoid tissue where the immune response is facilitated. Furthermore, the effect of belimumab treatment on T cell counts was not assessed; however, belimumab has been demonstrated to exert only mild effects on T cell populations, if any, and these appear to be restricted to the subset of CD8 + effector memory T cells.18 30 It is also pertinent to note that data may have differed between those initially enrolled into belimumab 1 mg vs 10 mg groups compared with placebo. The effect of the 76-week delay in the placebo group starting belimumab treatment was not formally investigated; however, B cell and IgG changes for placebo and active groups were similar (with regard to the timing and magnitude of the response) after accounting for the time shift,31 justifying the pooling of these groups in this analysis. Moreover, results from phase III studies showed similar reductions in multiple B cells and plasma B cell subsets with belimumab for patients who received 1 mg/kg dose to those in patients who received the 10 mg/kg dose.15 Furthermore, the patients enrolled in the continuation study were a subpopulation of the initial study, and of these, some did not have SRI-4 and biomarker data up to 312 weeks (6 years) available for analysis.
Conclusions
Belimumab treatment up to 284 (~5.5 years) and 312 weeks (6 years) resulted in substantial decreases in most of the assessed circulating B cell subset counts and in serum IgG levels. The observed decreases in B cell subsets and IgG levels with long-term belimumab treatment are in line with its mechanism of action and with observations from previous analyses of patients with SLE.13 15 Elevated baseline naïve B cells were associated with improvements in SRI-4, while elevated baseline SLE subset plasma and short-lived plasma B cells, and early increases in IgG levels, were associated with poorer SRI-4 response. Elevated baseline IgG titre was associated with significantly increased infection rates. Assessment of these biomarkers before initiating long-term belimumab treatment may have value in predicting response and infection risk and may also assist in counselling and management of patients with SLE.
Data availability statement
Data are available upon reasonable request. Anonymised individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and the study was performed in accordance with the principles of the Declaration of Helsinki. All sites (Schulman Associates Institutional Review Board; Biomedical Research Alliance of New York, LLC, Institutional Review Board; NYU School of Medicine Institutional Review Board; University of Kansas Medical CenterCentre - Human Subjects Committee; Oklahoma Medical Research Foundation - Institutional Review Board, Institutional Review Board - The Johns Hopkins University East Baltimore Campus [(Central Office]); University of Chicago Institutional Review Board; Western Institutional Review Board [(WIRB]); Ochsner Clinic Foundation Institutional Review Board; Wake Forest Baptist Health Institutional Review Board; Montefiore Medical Center, Internal Review Board; Cedars Sinai Medical Center Institutional Review Board; Gundersen Clinic Limited, Human Subjects Committee; University of Arizona Institutional Review Board; Medical University of South Carolina Office of Research Integrity; Office of Human Research Ethics, The University of North Carolina at Chapel Hill; MedStar Research Institute; Institutional Review Board, University Hospital of Brooklyn, Kings County Hospital; University of Michigan Medical School IRBMED; University of Pittsburgh Institutional Review Board; University of Southern California [(USC]) Health Sciences Campus Institutional Review Board) maintained ethics committee and institutional review board approval.
Acknowledgments
The authors thank Q2 Solutions Laboratories for conducting sample analysis of the B cell subset quantification, and Dr Thi-Sau Migone for her input in the study design and data analysis. Medical writing assistance was provided by Jennie McLean, PhD, of Fishawack Indicia Ltd, UK, part of Fishawack Health, and was funded by GlaxoSmithKline (GSK).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors DAR, WWF and WS were involved in study concept and/or design; WS was involved in acquisition of data; and DAR, HS, LE, MK, WWF and WS contributed to analysis and/or interpretation of data. HS is the guarantor. All authors reviewed the final draft of the manuscript and approved it for submission.
Funding Study BEL112233 (ClinicalTrials.gov identifier NCT00724867) and its BLISS-76 parent study (BEL110751; ClinicalTrials.gov identifier: NCT00410384) were funded by GSK.
Competing interests HS and DAR are employees of and own stocks/shares in GSK. MK and LE are former GSK employees and LE owns stocks/shares in GSK. WWF was an employee of HGS at the time of the BLISS-76 Trial and is a former GSK employee. WS has received research support from GSK and was supported in part by grants UL1TR001855 and UL1TR000130 from the National Center for Advancing Translational Science (NCATS) of the US National Institutes of Health.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.