Article Text

Abstract
Objective The role of neutrophils in driving pathogenic B cell responses in SLE is not fully understood. In this study, we explored the link between immune complex (IC)–driven neutrophil activation, the release of B cell pro-survival factor BAFF and B cell activation using SLE clinical samples.
Methods BAFF levels were analysed in serum samples from patients with SLE (n=60) and healthy controls (HCs, n=20) by ELISA and correlated with markers of neutrophil activation and circulating IC levels. Neutrophils were stimulated with RNP/IgG ICs and neutrophil activation, the release of BAFF, and neutrophil-mediated B cell responses were studied in vitro.
Results Levels of BAFF in patients with SLE were associated with markers of disease activity, including anti-dsDNA antibody titres (r=0.33, p<0.05), serum C3 levels (r=−0.57, p<0.001) and levels of circulating ICs (r=0.39, p<0.05). Stimulation of neutrophils from healthy individuals with RNP-ICs in vitro induced the release of BAFF (p<0.05), concomitant with formation of neutrophil extracellular traps (NETs) (p<0.05). In culture, neutrophils promoted B cell survival (p<0.05), proliferation (p<0.05) and CD27hiCD38hi plasmablast differentiation.
Conclusions Our results support a new mechanism by which ICs, on NET formation, induce the release of B cell pro-survival factor BAFF by neutrophils. Furthermore, neutrophils directly promoted B cell activation and cell differentiation. Targeting neutrophil–B cell interactions can be further explored as an approach for inhibiting pathogenic B cell responses in SLE.
- B-lymphocytes
- lupus erythematosus, systemic
- autoantibodies
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The levels of B cell activating factor (BAFF) are elevated in patients with SLE. However, the cellular sources of BAFF and the potential stimuli promoting BAFF release in SLE has not been fully explored.
What does this study add?
In vitro stimulation of neutrophils with SLE-relevant RNP/IgG immune complexes induces the release of BAFF. Neutrophil–B cell crosstalk directly promotes B cell survival and activation.
How might this impact on clinical practice or future development?
Approaches targeting interactions between neutrophils and B cells could help inhibit pathogenic B cell responses in patients with SLE.
Introduction
SLE is a severe, potentially life-threatening autoimmune disease associated with the production of pathogenic anti-nuclear autoantibodies (Abs) and the formation of immune complexes (ICs). The mechanisms driving the activation of autoreactive B cells in SLE are not fully understood. The levels of the B cell-activating factor (BAFF or BLyS), a member of TNF superfamily of cytokines, essential for B cell survival and responses to antigens (Ags), are increased in patients with SLE1–3 and have been shown to correlate with the production of pathogenic autoantibodies.3 4 Despite the clinical importance of abnormal BAFF production, the cellular sources of BAFF, driving pathogenic B cell responses in SLE, remain poorly explored.
Activation of neutrophils and the formation of neutrophil extracellular traps (NETs) have been implicated in SLE pathogenesis, particularly in the acceleration of tissue inflammation and vascular damage.5 6 Still, relatively little is known how neutrophils may contribute to B cell responses and auto-Ab production. In this study, we explored whether ICs contribute to the release of BAFF from neutrophils and the direct effects of neutrophil–B cell interactions on B cell survival and activation.
Methods
Study subjects
Patients with SLE (n=60) were recruited from the University of Washington Medical Center. All patients fulfilled the revised ACR criteria for SLE (online supplemental table 1). Healthy controls (HCs) (n=20) with no history of autoimmune diseases or current infections were used. Anti-dsDNA antibody titres, complement component 3 (C3) and complement component (C4) levels, were obtained from clinical records.
Supplemental material
Serum BAFF levels
Serum BAFF levels were measured by ELISA using Human BAFF/BLyS/ TNFSF13B Quantikine ELISA Kit (R&D Systems, Minneapolis, MN).
Immune complexes quantification
FcγRIIA internalisation, a bioassay for IC quantification, was analysed by flow cytometry as described.7
In vitro neutrophil activation and BAFF quantification
Neutrophils (1×106 cells/mL) were incubated in poly-L-lysine–coated tissue culture plates, left untreated or stimulated with 10 µg/mL RNP-ICs prepared as described previously7 for 1 and 3 hours. Supernatants were collected at each time point, and BAFF and other cytokines (IL-6, TNF- α, IL-21 and IL-10) were measured using LEGENDplex bead-based assay (BioLegend). NETs were detached and analysed as described.8
For the visualisation of BAFF, neutrophils were seeded on a 48-well plate pre-coated with 0.01% poly-L-lysine (Sigma) and fixed in 4% paraformaldehyde for 1 hour and permeabilised in 0.2% Triton for 10 min. BAFF was detected by anti-BAFF (D7I1U) Rabbit mAb (1:100, #19944S; Cell Signaling Technology), followed by incubation with Cy5 AffiniPure Donkey Anti-Rabbit IgG (H+L) at 1:100 (Jackson ImmunoResearch) and DAPI at 1 µg/mL. Cells were visualised with an immunofluorescence (IF) microscope (EVOS cell imaging system; Life Technology).
Neutrophil and B cell co-culture and flow cytometry
Isolated B cells (1×106 cells/mL) were labelled with Tag-it Violet (BioLegend) and cultured alone or co-cultured with autologous neutrophils (2.5×106 cells/mL) in RPMI 1640 containing L-glutamine and NaHCO3, supplemented with 10% FCS (Atlanta Biologicals), and 1% penicillin/streptomycin (Sigma), in the absence or presence of F (ab′) 2 anti-human IgM (anti-IgM) (10 µg /mL; Jackson ImmunoResearch) for 1–5 days. Cells were analysed by flow cytometry. Cell viability was measured by Zombie-NIR staining (Biolegend) and cell proliferation was measured by the dilution of cell-tracing dye. Analysis of B cell subsets was performed using a combination of fluorescently conjugated Abs (online supplemental table 2) by flow cytometry (Beckman Coulter, Brea, CA). Data were analysed using FlowJo (Tree Star, Ashland, OR).
Statistical analysis
Statistical tests were performed using Prism 8.0 (GraphPad, San Diego). Non-parametric Mann-Whitney U test and paired Student t-test were used to compare group values. Correlations were performed using Spearman’s correlation test. Results are reported as mean±SD. P values≤0.05 were considered statistically significant.
Results
BAFF titres in patients with SLE correlate with levels of circulating ICs
Consistent with prior work,1 3 4 9 BAFF levels were significantly higher in patients with SLE as compared with HCs, with approximately half of the patients with SLE having elevated levels of BAFF (figure 1A). The increased serum BAFF titres in patients with SLE correlated with anti-dsDNA Ab levels (online supplemental figure 1A), and inversely with levels of serum complement C3 and C4 levels (online supplemental figure 1B). The association between BAFF titres and the decrease of C3 and C4 levels was stronger in lupus nephritis patients’ subgroup (online supplemental figure 1C).
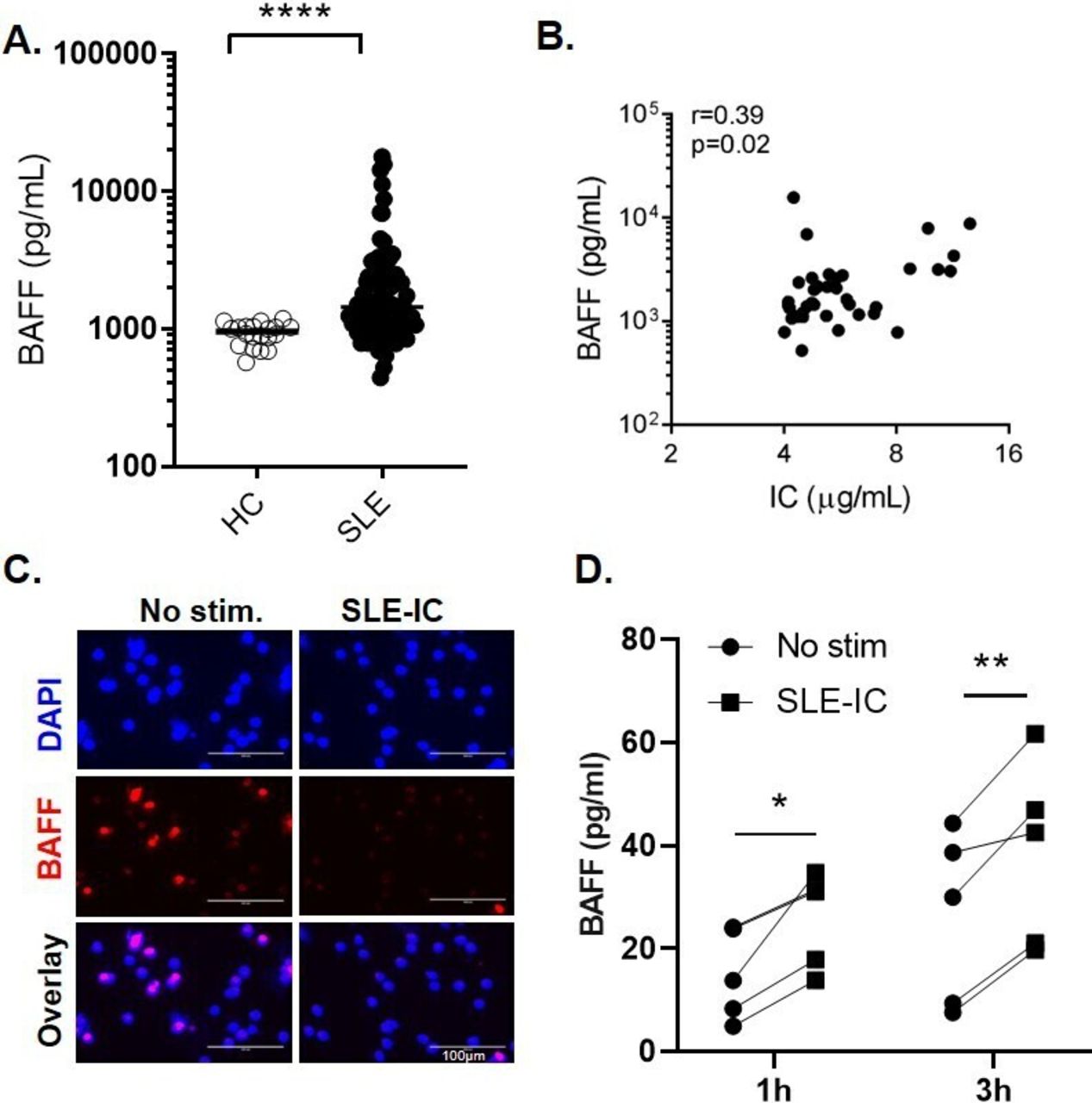
SLE immune complexes induce the release of BAFF from neutrophils. (A) Serum BAFF levels in patients with SLE (n=60) and healthy controls (HC) (n=20) measured by ELISA. Each symbol represents an individual; horizontal lines indicate mean. (B) Correlations between serum BAFF levels and circulating immune complexes (IC). (C–D) Neutrophils isolated from healthy controls (HC) were left untreated or stimulated with SLE-IC for 1 or 3 hours. (C) Immunofluorescent microscopic imaging showing nuclear DNA (blue) and BAFF (red) of neutrophils stimulated with SLE-IC for 1 hour. Data are representative of three independent experiments. Scale=100 µm. (D) BAFF release in cell supernatants in neutrophils from 5 individual HC (1 or 3 hours of cell culture), determined by ELISA. *p<0.05, **p<0.01 and ****p<0.0001, significance determined by Mann-Whitney U test or Student’s paired t-test, correlations determined by Spearman’s correlation.
Since complement C3 and C4 consumption has been linked to the presence of ICs,10 we explored the potential link between high levels of circulating ICs and levels of BAFF, using a novel flow cytometry–based bioassay.7 Levels of circulating ICs in patients with SLE correlated positively with levels of BAFF (r=0.39, p=0.02), suggesting that circulating ICs might be contributing to BAFF production (figure 1B).
In vitro stimulation of neutrophils with ICs induces the release of BAFF
Neutrophil activation by TLR ligands induces BAFF release.7 To investigate whether IC-driven neutrophil activation similarly can promote release of BAFF, we isolated neutrophils from patients with SLE or healthy individuals and stimulated them with SLE-ICs in vitro.
Consistent with prior work,7 RNP-IC could mediate a partial NET formation 1–3 hours after stimulation, as measured by the release of DNA (online supplemental figure 2). Next, we used a combination of immunofluorescent microscopy and ELISA to test whether the activated neutrophils release BAFF. Neutrophils stimulated with RNP-ICs showed a marked decrease in intracellular BAFF signal 1–3 hours after stimulation, as compared with unstimulated cells (figure 1C). The decrease in intracellular BAFF was associated with a significant increase in BAFF titres in cell culture supernatants (figure 1D), suggesting that, on IC stimulation, neutrophils release BAFF. Importantly, we observed a similar release of BAFF from neutrophils isolated from patients with SLE (p=0.0062 and p=0.042, at 1 hour and 3 hours, respectively) (online supplemental figure 3A).
The production of other cytokines, known to promote B cell activation, including IL-6 and TNF-α, was also increased after 3 hours of IC stimulation (although it did not reach statistical significance), while the levels of other cytokines, including IL-21 and IL-10 titres, remain unchanged (online supplemental figure 3B–E).
Neutrophils promote B cell survival and activation in vitro and drive SLE B cell differentiation
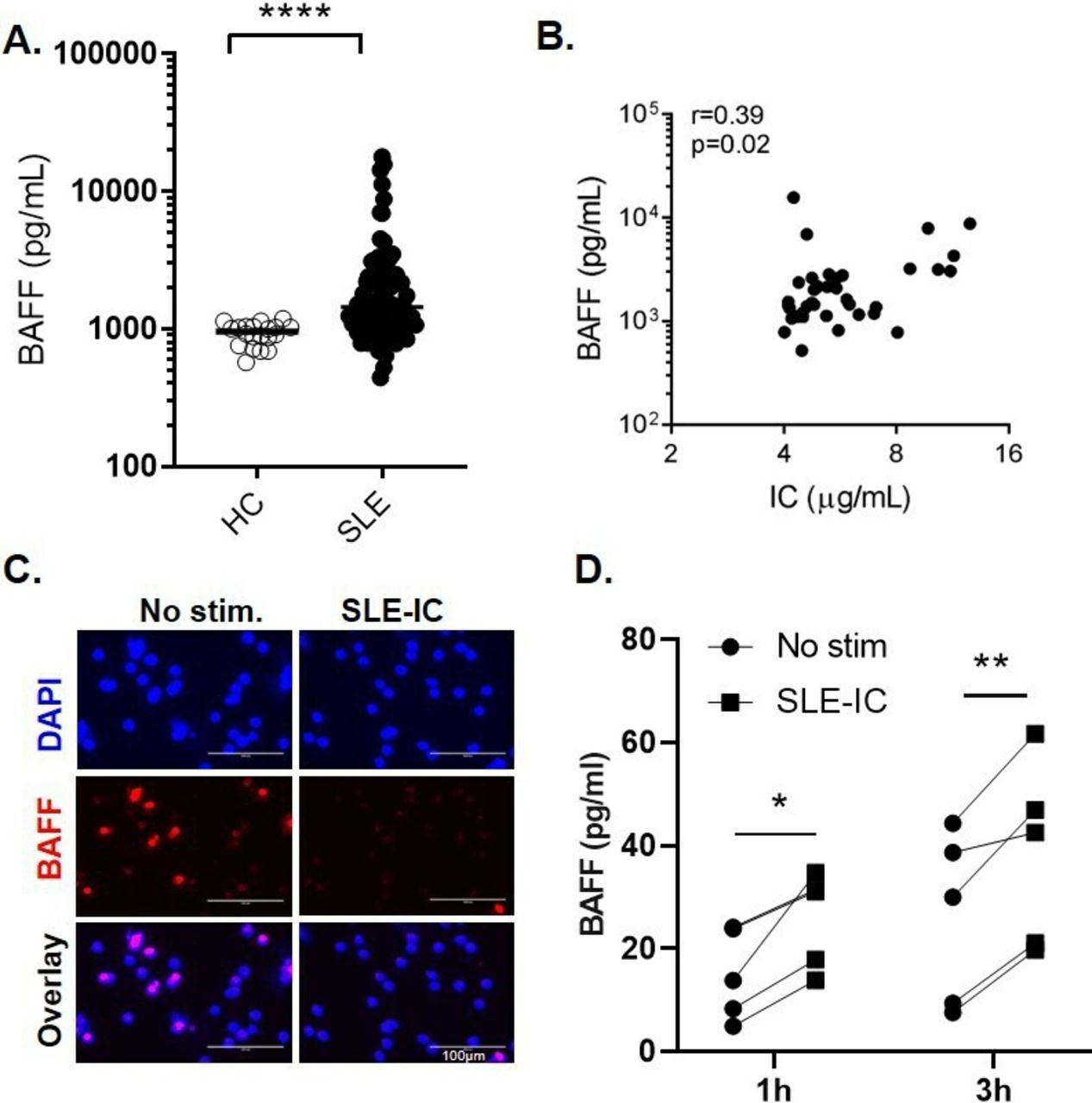
We next investigated the effects of neutrophils on B cell responses. We isolated CD19+ B cells from healthy individuals, and cultured them either alone or together with autologous neutrophils. After 5 days of incubation, the percentages of live B cells and proliferating B cells were higher in B cells–neutrophil co-cultures, as compared with B cells cultured alone, particularly, when cells were also stimulated with anti-IgM (figure 2A,B). The percentage of CD86+ (activated) B cells was also significantly increased in co-culture conditions without BCR crosslinking (figure 2C).

{kind=link}
{kind=link}
Co-culturing of B cells with neutrophils promotes B cell survival and activation. (A–C) B cells from HCs were labelled with Tag-it Violet and cultured alone (B cells), or co-cultured with autologous neutrophils (B cells/Nphs). B cells were left untreated (No stim.) or activated with 10 µg/mL anti-human F (ab′)2 IgM (anti-IgM). Cells were cultured for 5 days and then analysed by flow cytometry. (A) Representative flow analysis used to determine the frequencies of live B cells based on Fixable Viability Dye (FVD) staining. Gates depict the frequency of (FVD negative) live B cells. The graph below shows data from four independent experiments using different donors. (B) Representative flow analysis of cell proliferation using Tag-It Violet proliferation cell tracking dye; the graph below depicts the frequency of (Tag-it Violet) proliferating cells, graph below shows cumulative data from four independent experiments. (C) Representative flow analysis showing CD86 expression on gated B cells, gates depict the frequency of CD86+ B cells (gates defined based on unstained controls); the graph below shows cumulative data from four independent experiments. *p<0.05, by Student’s paired t-test. (D) B Cells isolated from PBMCs of patients with SLE were cultured alone or co-cultured together with autologous neutrophils for 5 days with or without anti-IgM stimulation and analysed by flow cytometry. Flow plots of gated live B cells of one SLE donor (representative of three independent experiments). Gates depict the frequency of CD27++ CD38++ (plasmablast) cells.
Using similar in vitro experimental settings, we also tested whether SLE neutrophils can also promote B cell differentiation into CD27hiCD38hi plasmablast. Notably, a distinct plasmablast population appeared on co-culturing of SLE B cells with neutrophils (figure 2D and online supplemental figure 4A–C). The frequencies of CD27hiCD38hi cells were increased by, but did not require BCR crosslinking, suggesting that neutrophil-derived signals alone were sufficient to drive their differentiation (figure 2D).
The effects of neutrophils on B cell differentiation were variable between patients with SLE. Analysis of several patients with SLE (n=5, online supplemental table 3) suggested that CD27hiCD38hi B cell differentiation in B cell–neutrophil co-cultures may depend on the presence of CD27hiCD38+/lo B cells, which disappeared in co-culture conditions, suggesting that they are the likely source of CD27hiCD38hi cells (online supplemental figure 4A). CD27hiCD38hi population showed a shift in CD86 expression and were larger in cell size as compared with CD27loCD38+/int naïve B cells and CD27++CD38+/lo memory B cells, further supporting their activated (blast-like) phenotype (online supplemental figure 4C).
Together, these data support the role of neutrophils in promoting B cell activation and their ability to directly drive SLE B cell differentiation into plasmablast.
Discussion
BAFF is an important B cell activating factor, involved in SLE pathogenesis and a target of the approved biologic belimumab. Consistent with prior work, we found elevated levels of BAFF in peripheral blood of patients with SLE associated with serological markers of disease activity, including complement consumption and anti-dsDNA antibodies.1 3 4 9
Although neutrophils are known to be an important source of BAFF, the potential stimuli promoting BAFF release in SLE had not been carefully explored. Given the serological findings of IC-driven disease associated with BAFF levels, as well as our prior work on IC-mediated NET formation, we hypothesised that ICs may be central in promoting BAFF release in SLE. Consistent with this hypothesis, prior studies have reported that exposure of neutrophils to bovine serum albumin-IC, after priming with G-CSF, induced the release of BAFF.11 Furthermore, Lood and Hughes showed BAFF release by neutrophils during NET formation.8
Our data revealed a reverse correlation between neutrophil counts and BAFF titres, and a positive association between increase in BAFF levels and levels of 8-OHdG DNA (oxidised DNA) and mt-COXII (mitochondrial) DNA (data not shown), further supporting the potential link between neutrophil activation/NET remnants and increased BAFF in patients with SLE.
To our knowledge, our study is the first to show that in vitro stimulation of neutrophils with disease-relevant RNP/IgG ICs induces the release of BAFF from human neutrophils. Our in vitro data suggest that this is likely an active process of BAFF release from intracellular storages, associated with neutrophil’s activation and NETosis. During this process, BAFF, released by the neutrophils, may also physically associate with the NETs.
Consistent with the release of BAFF, we found an increase of B cell survival in neutrophil/B cell co-cultures. Under the experimental conditions used, IC-stimulation did not promote significant release of other cytokines such as IL-21, IL-10, IL-6, TNF-α or CD40L, although we saw a trend towards the increased release of IL-6 (p=0.081) and TNF-α (p=0.2125) at 3 hours after IC stimulation, which may contribute to the effects of neutrophils on B cells. We also found an increase in the production of BAFF-related cytokine APRIL (data not shown); however, unlike BAFF, serum levels of APRIL did not correlate with IC levels, markers of neutrophil activation, or decrease of complement levels in patients with SLE.
Only a few published studies have explored the ability of neutrophils to activate B cells.2 12 13 Puga et al have reported on B-helper neutrophils in the splenic marginal zone, driving the activation of IgM-producing marginal zone B cells via the secretion of BAFF, APRIL and IL-21.12 More recently, Gestermann et al have shown that LL37–DNA complexes, present in NETs, can drive the activation of memory B cells and the production of anti-LL37 Abs.13 We propose that during NET formation, the extrusion of autoantigens, including LL37-DNA, oxidised mitochondrial DNA, and other mitochondrial components or neutrophil-specific enzymes,14 in combination with BAFF, could be a strong driver of autoreactive B cell responses in SLE. Despite the appearance of CD27hiCD38hi cells in co-culture conditions, we did not detect a significant anti-nuclear auto-Ab production. Different experimental conditions could explain the discrepancy of our data from Gestermann’s study. While Gestermann et al induced NET formation by anti-LL37 antibody to promote memory B cell activation and anti-DNA Ig production; in our study, we co-cultured SLE neutrophils and B cells without prior neutrophil activation. Our data, however, suggest that similar to Gestermann et al, CD27hiCD38hi cells derive from a subset of memory B cells, which are present in some, but not all, patients with SLE. Why SLE neutrophils selectively activate memory B cells, exactly what subset of memory B cells these cells correspond to, and what their Ag-specificity is, would need further in-depth analysis.
In brief, the results presented in this study support a novel mechanism in which ICs mediate NET formation and the release of BAFF. Consistent with the role of BAFF on B cells, our data support the ability of neutrophils to promote B cell activation and Ab-producing cell differentiation. Approaches to target the crosstalk between neutrophils and B cells could help inhibit pathogenic B cell responses in patients with SLE.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the University of Washington Institutional Review Board (IRB), with the reference ID STUDY00004145. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank patients with SLE and healthy volunteers for their contribution to this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TW and AV contributed equally.
Contributors TW, AV, JM, SSG and CL performed experiments, analysed data and interpreted results. TW and NVG designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Funding This work was funded by UW Division of Rheumatology start-up funds (NVG) and NIH T32 Research Training in Rheumatology (AV) and by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under Award Number UL1 TR002319 (ITHS Catalyst Award) (NVG and TW) and, in part, by a grant from the National Institutes of Health (R01AI44257) and Lupus Research Alliance Grant (519408) (NVG).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.