
Abstract
Surviving a single infection often results in lifelong immunity to the infecting pathogen. Such protection is mediated, in large part, by two main B cell memory ‘walls’ — namely, long-lived plasma cells and memory B cells. The cellular and molecular processes that drive the production of long-lived plasma cells and memory B cells are subjects of intensive research and have important implications for global health. Indeed, although nearly all vaccines in use today depend on their ability to induce B cell memory, we have not yet succeeded in developing vaccines for some of the world’s most deadly diseases, including AIDS and malaria. Here, we describe the two-phase process by which antigen drives the generation of long-lived plasma cells and memory B cells and highlight the challenges for successful vaccine development in each phase.
Similar content being viewed by others

Introduction
There are [pathogens] I’ll remember
All my life, though some have changed
— In My Life, Lennon–McCartney (1965)
An appreciation of the protective power conferred by immunological memory preceded our understanding of the cellular and molecular basis of this memory by centuries. During a plague in Athens in 430 B.C.E., the citizens understood that having the good fortune to recover from the disease, they could care for the newly stricken because no one was ‘attacked twice, at least not fatally’1. The simple principle that survival of an infection or exposure to a less virulent or attenuated form of a pathogen leads to lifelong immunity formed the foundation of Edward Jenner’s development of a vaccine for smallpox, an infectious disease that had the power to decimate entire populations in Europe in the mid-1700s2. Even fairly recent vaccines, such as those that elicit protective immunity to polio, measles and rubella, were developed for the most part without comprehensive knowledge of the mechanisms underlying immunity. Unfortunately, not all attempts to develop vaccines have been successful; worse, some vaccines were actually shown to be harmful to humans, as was the tragic case for respiratory syncytial virus3. In addition, to date, we have not succeeded in producing effective vaccines for some of the world’s most lethal diseases, including AIDS and malaria4,5. The hope is that more detailed knowledge of the cellular and molecular mechanisms that underlie the generation of protective, long-lived antibody responses and B cell memory will allow the development of safe, effective vaccines for pathogens for which we currently have none. Moreover, such knowledge may have broader benefits; for example, for the development of therapies for both systemic autoimmune disease and B cell tumours that may be, in part, unintended consequences of the drive to generate B cell memory6,7.
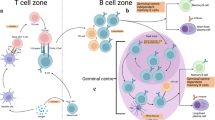
We now understand that immunological memory for many infectious diseases is acquired after a single infection and is dependent on the acquisition of two main ‘walls’ of memory; namely, long-lived plasma cells that produce protective antibodies and memory B cells that are able to respond on reinfection to pathogens and their variants. This process is detailed in several excellent recent reviews8,9,10,11. The antigen-driven generation of long-lived plasma cells and memory B cells from the naive B cell repertoire in the primary response to antigen occurs predominantly in secondary lymphoid organs (SLOs) in B cell follicles and in germinal centres (GCs) in two consecutive phases12,13,14. In phase 1, antigenic stimulation through B cell antigen receptors (BCRs) induces naive B cells to differentiate into short-lived plasma cells and GC B cells in the B cell follicles. In phase 2, antigens drive GC B cells to differentiate into long-lived plasma cells and memory B cells in GCs. In subsequent recall responses to antigens, memory B cells respond by differentiating into long-lived plasma cells or by re-entering the GC reaction. In this Review, we describe these two main phases of memory B cell development, highlighting some of the key variables that predict success at each step. We also briefly describe how chronic infectious diseases, including AIDS and malaria, may derail the acquisition of protective B cell memory. In this context, we speculate on the utility of leapfrogging over the development of vaccines for such chronic infectious diseases and providing protection through prophylactic, broadly neutralizing antibodies.
Phase 1 of B cell memory
Antigen-driven differentiation of naive B cells
Naive B cells first encounter antigen in the B cell follicles of SLOs15 (Fig. 1). Antigen binding to BCRs results in downstream BCR signalling and in the internalization, processing and presentation of the BCR-bound antigen on MHC class II molecules16. Antigen-activated naive B cells increase their metabolic activity17 and express chemoattractant receptors (CC-chemokine receptor 7 (CCR7) and EBI2) that direct them to the border of the T cell zone18, where they interact with antigen-specific T helper cells (TH cells) that have been primed by antigen-presenting dendritic cells (DCs) to differentiate towards T follicular helper cells (TFH cells)19 (Box 1). Recent studies suggest that during virus infections there may be additional layers of regulation of B cell responses at the follicular border mediated by IL-4-producing natural killer T cells20. Naive B cells appear to be multipotent and have at least three fates following interactions with TH cells. Naive B cells can differentiate into short-lived plasma cells that rapidly produce pathogen-specific antibodies of both switched and unswitched isotypes, albeit of relatively low affinity. Short-lived plasma cells accumulate in the red pulp regions of the spleen and medullary cords of lymph nodes, and their lifespans are generally limited to the course of the infection21. Naive B cells can also differentiate into GC B cells that will subsequently enter GCs in phase 2 and differentiate into long-lived plasma cells and memory B cells. Naive B cells may also give rise to memory B cells in the follicle independently of GCs. Indeed, B cells in BCL-6-deficient mice, which cannot develop GCs, were shown to differentiate into both IgM+ and IgG+ memory B cells that had not acquired somatic hypermutation (SHM), but these B cells did not differentiate into long-lived plasma cells22. Subsequent studies showed that GC-independent memory B cells that were mainly IgM positive were derived directly from a multipotential precursor cell early in the immune response and that these same precursors also gave rise to GC-dependent, switched immunoglobulin-positive memory B cells late in the response12. Lastly, long-lived memory B cells and both short-lived plasma cells and long-lived plasma cells can be generated by GC-independent and T cell-independent mechanisms21,23,24. T cell-independent type I and type II antigens have both been shown to generate long-lived plasma cells. Memory B cells were shown to be induced by T cell-independent type II polysaccharide antigens; however, these memory B cells show very low levels of SHM and isotype switching, are phenotypically distinct from memory B cells generated by naive B cells in response to T cell-dependent antigen and are stringently regulated in secondary responses by IgG antibodies specific for the immunizing antigen24. Importantly, T cell-independent type II polysaccharide vaccines such as Pneumovax confer long-term antibody protection in adult humans.

In phase 1 of the primary response (left), naive B cells exit the circulation, enter B cell follicles in the secondary lymphoid organ and survey the environment for antigen. Antigens encountered on follicular dendritic cells (FDCs) activate B cells through the B cell receptor (BCR), and the antigens are processed and presented to T cells at the T cell–B cell border, driving naive B cells to proliferate and differentiate into three main cell types: germinal centre (GC)-independent memory B cells, GC B cells or short-lived plasma cells. In phase 2 of the primary response (right), newly differentiated GC B cells form GCs and undergo proliferation and somatic hypermutation in the dark zone before exiting to the light zone, where the GC B cells encounter antigen on FDCs, present the antigen to T follicular helper cells (TFH cells) and undergo three main fates: namely, differentiation into memory B cells, differentiation into long-lived plasma cells or re-entry into the GC dark zone. In the secondary response (bottom right), memory B cells respond to antigen and differentiate into long-lived plasma cells or GC B cells that undergo GC reactions. TCR, T cell receptor.
The different antigen-driven T cell dependent fates have a profound impact on the outcome of an infection, resulting in either short-lived plasma cells that provide immediate short-lived antibody responses capable of controlling the initial infection or GC B cells that contribute to long-lived B cell memory by differentiating into long-lived plasma cells and GC-dependent memory B cells, albeit after a delay of several days to complete the GC reaction. A fundamental question for which we do not yet have a complete answer is what parameters govern this fate decision?
Precursory frequency, antigen affinity and avidity
A recent study provided important insights into the processes involved in antigen-driven naive B cell differentiation. The authors examined the factors that influence the outcome of vaccination in mice containing naive B cells expressing human germline VRC01-class BCRs that recognize the broadly neutralizing epitope of HIV gp120 (ref.25). Taking advantage of the remarkable array of tools available to determine the frequency and antigen affinity of VRC01-class B cells and detailed knowledge of the VRC01-class B cell antigen, this study provided evidence that the precursor frequency of naive B cells expressing germline VRC01-class BCR (~1 in 106), antigen affinity (less than 1 μM) and avidity (an antigenic epitope valency greater than 60) were interdependently limiting for successful GC completion. High-affinity multimeric antigens were shown to be capable of driving relatively rare VRC01-class naive B cells to differentiate into GC B cells that underwent extensive SHM and differentiated into memory B cells following a single immunization.
Recent studies of the transcriptional regulation of GC B cell differentiation versus plasma cell differentiation provided additional evidence for a role for antigen affinity in fate decisions. The transcription factor interferon regulatory factor 4 (IRF4), which regulates plasma cell differentiation, was shown to also initiate the generation of GC B cells, depending on the level of expression of IRF4, determined by the strength of BCR signalling26. Transient low levels of IRF4 favoured GC cell fates in contrast to sustained and higher levels of IRF4, which promoted the generation of plasma cells. These findings suggest that low-affinity antigen–BCR interactions that result in weak induction of IRF4 will initiate GC B cell differentiation, whereas high-affinity antigen–BCR interactions that induce high levels of IRF4 will promote plasma cell differentiation. TFH cell help provided both through CD40L-induced CD40 signalling and through IL-21 appear to play roles in directing naive B cells towards GC differentiation versus plasma cell differentiation27. However, chronic CD40 signalling antagonized the differentiation of GC B cells and promotes plasma cell differentiation28,29. Recently, evidence was provided that B cell fate decisions are made in a multistep process in which CD40 signalling is required only early to achieve increases in BCL-6 expression and that prolonged CD40 signalling drives precursors away from GC B cell fates30.
These observations raise an important question: how stringent is the influence of affinity of the BCR on the fate of naive B cells? Recent studies comparing the affinity thresholds for BCR-induced activation of human tonsil naive B cells versus GC B cells provided evidence that naive B cells have an intrinsic affinity threshold that is 1/100 of that of GC B cells31. The estimated affinity of the BCRs expressed by naive B cells for the antigen in this study was ~0.5 µM, similar to the suggested minimal affinity threshold for naive VRC01-class B cell selection in the study described earlier (~1 µM)25. Given that these affinities are in the lowest range of affinities of functional antibodies32 it appears that there may be very little affinity-dependent exclusion of antigen-specific naive B cells in the first phase of antigen-driven differentiation. Even relatively low-affinity antigen-specific naive B cells would enter GCs and have an opportunity to differentiate into high-affinity long-lived plasma cells and memory B cells. Thus, very few antigen-specific B cells may be ‘wasted’ in phase 1 as a high affinity for antigen is not essential.
Molecular mechanisms of fate instruction
Several recent studies addressed the molecular mechanisms that instruct B cells in plasma cell and GC fates in vivo, emphasizing the importance of both metabolic changes and TFH cell interactions. In one study, mice that contained B cells deficient in protein kinase Cβ (PKCβ), a key regulator of both B cell polarization and mechanistic target of rapamycin (mTOR) complex-dependent metabolic reprogramming, failed to develop plasma cells or GCs following immunization with a T cell-dependent antigen33. Following this, BCR signalling in naive B cells was shown to trigger a shift from canonical to non-canonical autophagy, and enhancement of the non-canonical pathway changed mitochondrial homeostasis and influenced the GC and plasma cell fate decisions34. In another study, mice containing B cells that were diminished in their ability to form long-term conjugates with T cells were severely impaired in their ability to form GCs and to produce affinity mature antibodies following immunization35. In this study, the diminished ability to form B cell–T cell conjugates was due to a deficiency in intersectin 2 (ITSN2), a guanine exchange factor for CDC42, a GTPase that is essential for the remodelling of the actin cytoskeleton in response to BCR signalling.
Impact of antigen structure
The nature and structure of antigens also impact the outcome of immunization. Of particular interest are complexes of weakly immunogenic recombinant subunit antigens coupled to bacterial membrane complexes, such as the outer membrane complex of Neisseria meningitidis36, or to virus-like particles, which are non-replicating shells composed of viral structural proteins that when overexpressed assemble into dense, multiprotein arrays37. These antigens are proving to be exceptionally immunogenic and capable of routinely inducing high-titred persistent antibody responses, although the mechanisms underlying their potency are not well understood. Indeed, the highly successful human papillomavirus (HPV) vaccine, which comprises virus-like particles assembled from the major L1 proteins from four HPV types, provides near-complete protection from sexually transmitted HPV infections in naive individuals and may even be effective when administered in a single dose38. The HPV vaccine was shown to induce memory B cells in vaccinated naive individuals expressing potently neutralizing antibodies but ones that have not undergone extensive SHM39.
Another important feature of antigens is the form they take on in vivo. Antigens arrive in the SLO through afferent lymphatics via subcapsular sinuses. LYVE1-expressing lymphoid endothelial cells and a network of surrounding CD169-expressing macrophages create a tight barrier that prevents movement of large antigens and antigen complexes into B cell follicles40. However, small antigens such as hen egg white lysozyme (HEL; 14.4 kDa) have been shown to penetrate B cell follicles within minutes after injection and pass into pores of the subcapsular sinuses or lymphatic conduits41,42. Larger antigens (more than 70 kDa), viruses and virus-like particles are captured by subcapsular sinus macrophages in the form of immune complexes or complement-coupled complexes. These are rapidly shuttled to non-cognate B cells that transport the antigens into follicles, where the complexes are deposited onto follicular DCs (FDCs)40,43 that provide a continuous supply of antigen during the B cell response44. FDCs also provide a 3D network on which B cells move within the follicle and secrete CXC-chemokine ligand 13 (CXCL13), which recruits and retains CXC-chemokine receptor 5 (CXCR5)-expressing B cells in GCs44,45,46. The fact that in SLOs, B cells are exposed to both soluble and FDC-associated antigens raises an important question: is the outcome of a B cell’s exposure to soluble antigens versus membrane-associated antigens similar? It is well established that membrane-associated antigens are highly effective at triggering B cell activation47. In addition, several studies in vitro have provided evidence that the requirements for B cell responses to soluble antigens versus membrane-associated antigens differ in a variety of parameters, including the requirement for co-receptors, responses to monovalent antigens, the regulation of cytoskeleton and the kinetics and regulation of BCR clustering31,48,49. However, at present we have little information concerning the fate of B cells responding to soluble antigens versus membrane-associated antigens either in vitro or in vivo. It would seem that vaccine design would benefit from such information.
Effect of pathogen products in the microenvironment
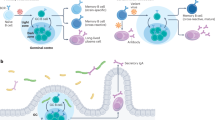
The impact of the microenvironment within the B cell follicle on the fate of naive B cells is just beginning to be explored. One important environmental factor is the presence of the pathogen or pathogen products and the signals of imminent danger these send to the host through innate immune receptors. A recent study showed that pathogen-associated molecular patterns (PAMPs), in particular the CpG ligand for Toll-like receptor 9 (TLR9), had the unexpected effect on naive B cells of blocking antigen processing and presentation at a point after antigen internalization but before the delivery of the antigen to processing compartments50. Thus, CpG-stimulated B cells were less able to acquire T cell help. Parallel studies provided evidence that activated B cells that were unable to acquire T cell help shortly after BCR signalling underwent apoptosis due to induced mitochondrial dysfunction acting as a ‘metabolic clock’17. These studies also showed that TLR9 stimulation rescued the antigen-activated B cells from apoptosis and drove their proliferation and differentiation into low-affinity short-lived plasma cells. Taken together these observations provide a mechanism by which naive B cells activated in the presence of pathogen products are fated to rapidly differentiate into short-lived plasma cells rather than committing to participate in time-consuming GC reactions (Fig. 2).

Naive B cells express both B cell receptors (BCRs) for antigen and Toll-like receptors (TLRs; depicted as the intracellular TLRs) that respond to pathogen-associated molecular patterns (PAMPs). In the presence of antigen alone (left), B cells internalize, process and present antigen on MHC class II molecules. Engagement with antigen-specific preactivated T cells results in B cell proliferation and differentiation into germinal centre (GC) B cells. In the presence of antigen and PAMPs (right), signalling via TLRs blocks the processing of antigens internalized by BCRs and stimulates B cells to proliferate and differentiate into short-lived plasma cells. TCR, T cell receptor.
The discovery of an antagonistic effect of CpG on B cell responses to a T cell dependent antigen was surprising as responses to immunization with T cell-dependent antigen are well known to require adjuvants, many of which contain PAMPs as immunostimulatory components. Conventional DCs respond to PAMPs in peripheral tissues, where they encounter antigen, by increasing their antigen-presenting cell function before travelling to SLOs to activate T cells, a clear benefit of adjuvants. Indeed, the yellow fever vaccine YF-17D, one of the most successful empiric vaccines ever developed, activates DCs through multiple TLRs51,52. However, the study just described suggests that if PAMPs accumulate in B cell follicles, B cells will respond to the signal of impending danger by differentiating immediately into relatively low-affinity short-lived plasma cells. That study showed that immunization of mice and humans with protein antigens with CpG as an adjuvant resulted in high levels of antibody that failed to affinity mature, consistent with GC-independent plasma cell generation50. These observations suggest that PAMPs or CpG in particular may not be ideal adjuvants for pathogen-specific responses that require high levels of SHM, such as broadly neutralizing HIV-specific antibody responses.
Are there better adjuvant formulations to drive naive B cells to differentiate into GC B cells that would accumulate SHM in GCs? A recent comparison of the impact of eight different commonly used adjuvants on the antibody response to the HIV envelope protein in a non-human primate model showed that formulation of the envelope protein with adjuvants increased the levels of HIV-specific antibodies but did not increase the frequency of SHMs essential for the development of broadly neutralizing antibodies53. Thus, future evaluations of the efficacy of adjuvants may require an assessment of not only antibody titres and the duration of the response but also of evidence for stimulation of GC responses leading to SHM and affinity maturation.
Phase 2 of B cell memory
Antigen-driven differentiation of GC B cells
In contrast to the first phase of memory generation, in which, as previously described, antigen affinity selection appears to play a minimal role in fate decisions, the second phase of memory generation that occurs in GCs is mostly about producing high-affinity B cells through selection (Fig. 1). In this phase, GC B cells differentiated from naive B cells enter newly forming GCs, which are tightly confined clusters of cells within the follicle. Here, the GC B cells highly express both the transcription factor BCL-6 and the G-protein-coupled receptor sphingosine 1-phosphate receptor 2 (S1P2), which promotes their retention in the GC54,55,56. In the GC dark zone, GC B cells proliferate and undergo SHM before exiting to enter the GC light zone, where they are selected to undergo one of three fates. First, some GC B cells differentiate into long-lived plasma cells that take up residence in the bone marrow and actively secrete large quantities of antibodies, persisting for years, potentially a lifetime, in the absence of antigen or a secondary challenge21,57. Second, some GC B cells differentiate into long-lived memory B cells, which are quiescent cells that reside in niches within SLOs and other tissues that promote their exposure to antigen. Third, GC B cells may re-enter the GC dark zone for additional rounds of SHM and selection, ultimately giving rise to memory B cells or long-lived plasma cells17,58,59,60. In secondary responses, on antigen rechallenge, memory B cells are activated and have at least two fates: they either differentiate into long-lived plasma cells or enter GCs to undergo rounds of population expansion, SHM and selection (Fig. 1).
Affinity thresholds and GC B cell selection
To understand affinity selection in GCs, the mechanisms by which B cells sense their affinity for antigens is of central importance. As mentioned earlier, both soluble antigens and membrane-associated antigens may be present in GCs. At present, the mechanisms by which B cells sense their affinity for soluble antigens are only poorly understood. In contrast, the ability of B cells to engage antigen, signal in response to it and internalize it for processing and presentation were shown to be directly dependent on the B cells’ affinity for the antigen16,61,62. B cells capture antigen from membranes through the exertion of pulling forces on the BCR–antigen complex. Higher-affinity interactions are better able to resist the pulling forces and are more successful at internalizing antigen for presentation to TFH cells. Thus, B cell affinity is indirectly determined by the amount of antigen a B cell is able to capture from an FDC and present to a TFH cell in the GC (Fig. 3).

On antigen engagement (step 1) germinal centre (GC) B cells form unique actin- and ezrin-rich pod-like structures that concentrate B cell receptors (BCRs) (step 2). GC B cells exert force on BCR–antigen complexes through myosin-dependent processes (step 3), leading to the deformation of the antigen-associated membrane (steps 4, 5) and ultimately to the capture of the antigen and associated membrane fragments (step 6) and their trafficking away from the synapse for internalization (step 7). High-affinity BCRs are better able than low-affinity BCRs to resist the pulling force exerted by the GC B cell on the BCR–antigen complex and, consequently, high-affinity BCRs capture, process and present larger amounts of antigen to T follicular helper cells.
If affinity selection is paramount in the GC reaction, are GC B cells particularly well suited to discriminate antigen affinity and to be selected by high-affinity antigens? As mentioned earlier, recent studies provided evidence that the intrinsic affinity threshold of GC B cells for both BCR signalling and antigen internalization is at least 100-fold higher than that of naive B cells31. Recent studies also provided evidence that GC B cells have ‘rewired’ CD40 and BCR signalling such that CD40 signals are transduced through nuclear factor-κB and BCR signals are transduced through forkhead box protein O1 (FOXO1), and both of these signalling pathways are required for induction of MYC, which is critical for GC B cell survival63. In contrast, naive B cells can signal through either BCR or CD40 alone to activate MYC. The net result of this rewiring is that, compared with naive B cells, GC B cells show a more stringent requirement for antigen and T cells for them to become activated.
In addition, GC and naive B cells differ in their expression of cell surface receptors that may serve to further increase the affinity threshold of GC B cells. For example, compared with naive human B cells, human GC B cells express very little VLA4 (also known as α4 integrin), which is an integrin that stabilizes the initial interactions with antigen-bearing DCs by binding to VCAM1 and thereby lowers the affinity threshold for B cell activation31. In addition, compared with naive B cells, GC B cells were shown in mice to recognize antigen through specialized immune synapse architecture48. Subsequent studies provided a detailed picture of the GC B cell architecture, showing that BCRs expressed by GC B cells are concentrated in unique, highly dynamic actin- and ezrin-containing pod-like structures through which the BCRs exert pulling forces and test affinity31. Remarkably, the affinity of the antigen engaged by the GC B cell dictates the behaviour of the pod-like structures. Low-affinity antigens induce a dynamic searching behaviour with engagement and release of the antigen, whereas high-affinity antigens induce stable long-lived engagement of the GC B cell with the antigen-containing membranes31. A more detailed understanding of the mechanisms underlying the initiation of BCR signalling in these GC B cell pod-like structures may provide insight into the design of vaccines to maximize selection of high-affinity GC B cells.
Further differentiation of GC B cells
A central question for which we are just beginning to get answers concerns the control of the differentiation of GC B cells into long-lived plasma cells versus memory B cells in GCs. At present, the data support the concept of a fundamental dichotomy between the GC processes that drive plasma cell differentiation versus memory B cell differentiation. Studies using 5-bromodeoxyuridine pulse labelling provided evidence that GC responses undergo a temporal switch as they mature, first producing memory B cells with limited SHM and then generating long-lived plasma cells that contain more highly mutated variable (V) genes14. The observed paucity of mutations in the memory B cell compartment suggests that B cells have lower levels of affinity-based selection and have more broadly cross-reactive BCRs as compared with more highly mutated BCRs of long-lived plasma cells. Recent studies directly demonstrated that affinity selection is not equally applied to precursors of memory B cells and long-lived plasma cells, resulting in highly selected, high-affinity long-lived plasma cells and broadly reactive lower-affinity memory B cells. Analyses of single nitrophenyl-specific plasma cells and memory B cells provided evidence that the initial output of GCs in response to immunization was stringently selected, high-affinity plasma cells64. In contrast, only 65% of memory B cells were capable of producing high-affinity antibodies, indicating less stringent selection. Consistent with these observations, additional studies showed that only GC B cells that had acquired high-affinity BCRs for the immunizing antigen HEL through SHM differentiated into plasma cells65. Subsequent studies provided additional mechanistic detail concerning the selection of GC B cells into the plasma cell lineage, showing that plasma cell lineage commitment was favoured by BCL-6lowCD69hi GC B cells that expressed the transcription factor IRF4 (ref.66). The generation of BCL-6lowCD69hi GC B cells, which also expressed high levels of ICAM1 and SLAM, was regulated by T cell-mediated signalling through CD40, suggesting that TFH cell–GC B cell interactions were key to the generation of GC B cells that were prone to differentiate into long-lived plasma cells.
Additional details concerning memory B cell differentiation were provided by studies showing that CCR6 uniquely marked memory B cell precursors in GC light zones and that these memory B cell precursors expressed predominantly low-affinity HEL-specific BCRs67. Studies using a transgenic mouse model in which GC B cells were inducibly marked showed that light zone B cells with lower-affinity BCRs were more prone to enter the memory B cell pool68. These memory B cell-prone B cells expressed higher levels of the transcriptional repressor BACH2, and their levels of BACH2 expression were inversely correlated with the strength of help provided by T cells. These data supported an instructive model for the selection of memory B cells in which T cells determine BACH2 expression, promoting differentiation to memory B cells, perhaps by promoting the transition of memory B cells to quiescence through the antiproliferative or prosurvival function of BACH2 (ref.68).
As memory B cells provide a second level of protection against re-exposure to pathogens by differentiating into either plasma cells or GC B cells, recent studies focused on determining if both fates reside in all memory B cells. Studies using a mouse model in which B cells expressing activation-induced cytidine deaminase (AID) were labelled, indicating that they had engaged in a GC reaction, showed that immunization with sheep red blood cells (a particulate antigen) resulted in the generation of several memory B cell subpopulations with distinct functions69. These included IgM+ and IgG+ B cells that persisted in GC-like structures for several months and IgM+ and IgG+ memory B cells that accumulated outside the follicle. On antigen challenge, the IgM+ memory B cells entered GCs in contrast to IgG+ memory B cells, which differentiated into plasma cells. In a transgenic mouse model, IgG1+ memory B cells were shown to be predisposed to differentiate into plasma cells and were the major source of IgG1 antigen-specific antibody in the secondary response70.
The fate of antigen-specific B cells that expressed IgM (IgM+) or isotype-switched immunoglobulin (swIg+) in C57BL/6 mice was traced following immunization in a separate study and it was found that both IgM+ and swIg+ memory B cells were generated71. However, in subsequent challenges, although IgM+ memory B cells were more numerous and longer-lived than swIg+ memory B cells, swIg+ memory B cells dominated the challenge response, producing plasma cells and new memory B cells but no GC B cells71. As antigen-specific serum immunoglobulin levels dropped, the numbers of swIg+ memory B cells declined but IgM+ memory B cells, which contained few SHMs but had the ability to produce GC B cells, persisted and became the reservoirs of durable memory. However, a subsequent study by the same group of authors suggested an alternative mechanism for the instability of swIg+ memory B cells and the generation of large numbers of IgM memory B cells in C57BL/6 mice, namely that the response in C57BL/6 mice was dominated by an unusual single heavy chain variable domain (VH) that conferred high-affinity binding to antigen72.
Another study provided evidence for functionally distinct memory B cell subsets on the basis of their expression of CD80 and PDL2, independently of isotype73. On challenge, CD80+PDL2+ memory B cells that were of relatively high affinity differentiated into plasma cells but not GC B cells, and conversely CD80–PDL2– memory B cells that were of lower affinity produced few plasma cells but robustly differentiated into GC B cells. In a separate study, CD80hi memory B cells were shown to be generated from high-affinity B cells during the primary immune response by mechanisms that depended on TFH cell-inducing strong CD40 signalling in contrast to the development CD80low B cells, which did not require strong CD40 signalling74.
Taken together, these studies provide strong evidence that the ability of memory B cells to differentiate into plasma cells versus GC B cells is compartmentalized into perhaps several subpopulations. To exploit this compartmentalization of memory B cell functions in vaccine design it will be necessary to further elucidate the mechanisms that drive the differentiation of these memory B cell subsets.
Roles of long-lived plasma cells and memory B cells in protective immunity
The current model of B cell memory suggest that the highly-selected, high-affinity antibodies produced by long-lived plasma cells form the first line of defence against homologous challenge and that memory B cells provide a second layer of defence against challenge by variant pathogens that escape the long-lived plasma cell-mediated defence (Fig. 4). How strong is the evidence that memory B cells function in this way and is the ability of memory B cells to provide this function dependent on accumulating additional SHMs?

Long-lived plasma cells in the bone marrow secrete highly selected, highly specific antibodies (depicted in red) that form a first ‘wall’ (bottom) against reinfection by homologous pathogens. Variant pathogens can find holes in this wall; however, having escaped the antibodies from the long-lived plasma cells, the variant pathogens encounter a second wall (top) formed by memory B cells that were less highly selected and therefore maintain a broader range of antigen affinities and specificities. The memory B cells are activated by the variant pathogen to differentiate into long-lived plasma cells or to re-enter the geminal centres (GCs) to replenish the memory B cell pool.
Studies using a mouse model of West Nile virus infection with wild-type and variant viruses that differed in only one amino acid in a dominant neutralizing epitope demonstrated that the antibodies produced by long-lived plasma cells generated in response to the wild-type virus only poorly neutralized the variant virus75. However, memory B cell-derived plasma cells produced antibodies that recognized both the wild-type virus and the variant virus equivalently, or remarkably, recognized the variant virus better than the wild-type virus and did so without accumulating additional SHMs. Consistent with these results, studies that traced influenza virus haemagglutinin (HA)-specific B cells in mice immunized first with the Narita virus strain followed by challenge with the homologous virus or the heterologous PR8 virus strain demonstrated that pre-existing antibodies secreted by long-lived plasma cells protected against homologous challenge, whereas protection from heterologous challenge required memory B cell activation. These memory B cells were primarily directed towards the relatively invariant HA stem76. Studies of the antibody response in humans to vaccination with the pandemic 2009 H1N1 influenza vaccine showed that individuals who had low levels of pre-existing antibodies to the vaccine generated broadly reactive antibodies to the HA stem, whereas high pre-existing levels of antibody to the vaccine correlated with strain-specific, HA-head responses, suggesting that antibodies to the HA head blocked the generation of broadly protective stalk-specific antibodies77. Thus, an individual’s immune history with influenza virus affects the ability to produce broadly protective B cell responses on challenge.
Of interest was the finding in mice that administration of the mTOR inhibitor, rapamycin, during immunization with influenza virus subtype H3N2 reduced the formation of GCs and inhibited class switching by B cells but resulted in a unique repertoire of antibodies that protected against lethal infection with heterosubtypic H5N1 virus78. Studies of the antibody response of mice immunized with Dengue virus envelope proteins and challenged with the same or variant virus proteins showed that the variant proteins stimulate predominantly IgM+ memory B cells with the most diverse and least mutated V genes79. Taken together, these studies provide strong evidence for the role of highly diverse memory B cell populations in providing broad protection against variant virus infections. The challenge for the future will be to determine how to differentially evoke these memory B cell populations.
Effect of chronic infections on memory B cells
Nearly all of the studies described thus far in this Review investigated the generation and function of B cell memory in models of acute infections or vaccinations followed by challenge. The findings from these studies are most relevant to antibody-mediated development of vaccines to induce protective immunity in naive individuals. However, there is an urgent need for vaccines for individuals who have chronic infections, including AIDS, malaria and hepatitis virus infections. The development of such vaccines may be highly challenging as current evidence indicates that such chronic infections have a profound impact on the memory B cell compartment, the repercussions of which we are just beginning to understand80. For example, one common feature of chronic infections is the large expansion of a novel population of T-bet+ memory B cells termed ‘atypical memory B cells’. We do not yet understand the function of these memory B cells in humans, and it is not entirely clear whether mouse models of T-bet+ memory B cells that arise with age and in certain infections are relevant to human atypical memory B cells and can be used as models to study atypical memory B cell80. Thus, we may be quite a distance from designing vaccines for chronic infections.
Prophylactic antibodies
Although vaccination to induce B cell memory is the most effective and low-cost method of preventing infectious diseases, the development of effective vaccines for many of the world’s most deadly pathogens, including those that cause AIDS and malaria, have proven to be extremely challenging and thus far have met with little success. The failure to develop vaccines for these two diseases as well as several others have led to efforts to leapfrog over vaccine development entirely and to directly provide highly effective broadly neutralizing antibodies as prophylactics (reviewed in refs4,5). This is not a new idea as passively transferred antibodies have been used for more than a century as therapeutics for infectious diseases such as diphtheria and tetanus and subsequently for several viral diseases, including infections with respiratory syncytial virus, hepatitis B virus and hepatitis C virus. What is new, however, is the successful generation of rare, highly potent and broadly cross-reactive human monoclonal antibodies for several viruses, including HIV, and for the parasite that causes malaria. In addition, antibody engineering can increase the potency, cross-reactivity and half-life of such antibodies. Key to the discovery of these rare human antibodies was the careful selection of donors with desirable serum antibody profiles and the development of high-throughput human B cell isolation technologies. At present, passive antibody prophylaxis appears to be a promising alternative to vaccination for HIV infection. In addition, recent successes in vector-mediated antibody transfers in mice and macaques in which a single injection provided continuous and sustained delivery of antibody may provide an alternative form of antibody prophylaxis55,56. A number of antiviral and antimalaria monoclonal antibodies are in clinical development, and the results of these studies over the next several years will tell whether the current promise of this approach is fulfilled.
Summary
We have come a long way towards providing the plague survivors of 430 B.C.E. Athens with an explanation of their good fortune to never be ‘attacked twice’. We now have a good understanding of the cellular basis of B cell memory, namely long-lived plasma cells and memory B cells, and of how these cells develop from naive B cells in two consecutive antigen-driven processes. We also have a general concept of how the two B cell memory walls are built and the protection against invading pathogens they afford. What we know far less about is how to effectively design vaccines and adjuvants to reproducibly generate B cell memory. At present, it is not clear if general guidelines will emerge from current research or if vaccine design will continue to be basically an empirical process. Given the recent rapid pace of progress in understanding B cell memory, we are optimistic that it will not be long before future advances provide new prescriptions for the development of badly needed vaccines to protect against humankind’s worst enemies.
References
Thukydides, Crawley, R. & Gavorse, J. The Complete Writings of Thucydides. The Unabridged Crawley Translation (The Modern Library, 1934).
Jenner, E. An Inquiry Into the Causes and Effects of Variolae Vaccinae (Sampson Low, 1798).
Kapikian, A. Z., Mitchell, R. H., Chanock, R. M., Shvedoff, R. A. & Stewart, C. E. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am. J. Epidemiol. 89, 405–421 (1969).
Walker, L. M. & Burton, D. R. Passive immunotherapy of viral infections: ‘super-antibodies’ enter the fray. Nat. Rev. Immunol. 18, 297–308 (2018).
Cockburn, I. A. & Seder, R. A. Malaria prevention: from immunological concepts to effective vaccines and protective antibodies. Nat. Immunol. 19, 1199–1211 (2018).
Mlynarczyk, C., Fontan, L. & Melnick, A. Germinal center-derived lymphomas: the darkest side of humoral immunity. Immunol. Rev. 288, 214–239 (2019).
DeFranco, A. L. Germinal centers and autoimmune disease in humans and mice. Immunol. Cell Biol. 94, 918–924 (2016).
Weisel, F. & Shlomchik, M. Memory B cells of mice and humans. Annu. Rev. Immunol. 35, 255–284 (2017).
Phan, T. G. & Tangye, S. G. Memory B cells: total recall. Curr. Opin. Immunol. 45, 132–140 (2017).
Kurosaki, T., Kometani, K. & Ise, W. Memory B cells. Nat. Rev. Immunol. 15, 149–159 (2015).
Cyster, J. G. & Allen, C. D. C. B cell responses: cell interaction dynamics and decisions. Cell 177, 524–540 (2019).
Taylor, J. J., Pape, K. A. & Jenkins, M. K. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med. 209, 597–606 (2012).
Ruddle, N. H. & Akirav, E. M. Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J. Immunol. 183, 2205–2212 (2009).
Weisel, F. J., Zuccarino-Catania, G. V., Chikina, M. & Shlomchik, M. J. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 44, 116–130 (2016). This study reveals that memory B cells and long-lived plasma cells are produced in different phases of the GC reaction, with memory B cell formation preceding the formation of plasma cells.
Phan, T. G., Gray, E. E. & Cyster, J. G. The microanatomy of B cell activation. Curr. Opin. Immunol. 21, 258–265 (2009).
Kwak, K., Akkaya, M. & Pierce, S. K. B cell signaling in context. Nat. Immunol. 20, 963–969 (2019).
Akkaya, M. & Pierce, S. K. From zero to sixty and back to zero again: the metabolic life of B cells. Curr. Opin. Immunol. 57, 1–7 (2019).
Cyster, J. G., Dang, E. V., Reboldi, A. & Yi, T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol. 14, 731–743 (2014).
Garside, P. et al. Visualization of specific B and T lymphocyte interactions in the lymph node. Science 281, 96–99 (1998).
Gaya, M. et al. Initiation of antiviral B cell immunity relies on innate signals from spatially positioned NKT cells. Cell 172, 517–533.e20 (2018). This study shows that natural killer T cells, positioned in interfollicular areas of lymph nodes, secrete IL-4 in response to viral infections and this plays a critical role in initiating B cell responses.
Bortnick, A. & Allman, D. What is and what should always have been: long-lived plasma cells induced by T cell-independent antigens. J. Immunol. 190, 5913–5918 (2013).
Toyama, H. et al. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 17, 329–339 (2002).
Defrance, T., Taillardet, M. & Genestier, L. T cell-independent B cell memory. Curr. Opin. Immunol. 23, 330–336 (2011).
Obukhanych, T. V. & Nussenzweig, M. C. T-independent type II immune responses generate memory B cells. J. Exp. Med. 203, 305–310 (2006). This study shows T cell-independent antigens can generate memory B cells that are phenotypically different from T cell-dependent memory B cells but that can last for months after immunization.
Abbott, R. K. et al. Precursor frequency and affinity determine B cell competitive fitness in germinal centers, tested with germline-targeting HIV vaccine immunogens. Immunity 48, 133–146.e36 (2018).
Ochiai, K. et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38, 918–929 (2013). This study reveals that the transcription factor IRF4 plays a critical role in B cell fate decision, with its transient expression inducing GCs, whereas prolonged expression induces plasma cell differentiation.
Zotos, D. et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 207, 365–378 (2010).
Bolduc, A. et al. Constitutive CD40L expression on B cells prematurely terminates germinal center response and leads to augmented plasma cell production in T cell areas. J. Immunol. 185, 220–230 (2010).
Kishi, Y. et al. Augmented antibody response with premature germinal center regression in CD40L transgenic mice. J. Immunol. 185, 211–219 (2010).
Zhang, T. T. et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. eLife 6, e19552 (2017).
Kwak, K. et al. Intrinsic properties of human germinal center B cells set antigen affinity thresholds. Sci. Immunol. 3, eaau6598 (2018). This study shows that human GC B cells make cytoskeletal rearrangements and form actin- and ezrin-rich pod-like extensions to test their affinity against membrane-bound antigens.
Karush, F. in Immunoglobulins (eds Gary W. Litman & Robert A. Good) 85–116 (Springer, 1978).
Tsui, C. et al. Protein kinase C-beta dictates B cell fate by regulating mitochondrial remodeling, metabolic reprogramming, and heme biosynthesis. Immunity 48, 1144–1159.e45 (2018).
Martinez-Martin, N. et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science 355, 641–647 (2017).
Burbage, M. et al. Tuning of in vivo cognate B-T cell interactions by intersectin 2 is required for effective anti-viral B cell immunity. eLife 7, e26556 (2018).
Wu, Y. et al. Sustained high-titer antibody responses induced by conjugating a malarial vaccine candidate to outer-membrane protein complex. Proc. Natl Acad. Sci. USA 103, 18243–18248 (2006).
Bachmann, M. F. & Jennings, G. T. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 10, 787–796 (2010).
Schiller, J. & Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 36, 4768–4773 (2018).
Scherer, E. M. et al. Characteristics of memory B cells elicited by a highly efficacious HPV vaccine in subjects with no pre-existing immunity. PLOS Pathog. 10, e1004461 (2014).
Phan, T. G., Green, J. A., Gray, E. E., Xu, Y. & Cyster, J. G. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat. Immunol. 10, 786–793 (2009).
Pape, K. A., Catron, D. M., Itano, A. A. & Jenkins, M. K. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 26, 491–502 (2007).
Roozendaal, R. et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 30, 264–276 (2009).
Junt, T. et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450, 110–114 (2007). This study shows that the CD11b +CD169 +MHC class II + macrophage population that resides on the floor of subcapsular sinuses is responsible for capturing viral particles and presenting them to the underlying B cells to initiate the humoral response and to limit the systemic dissemination of pathogens.
Gonzalez, S. F., Pitcher, L. A., Mempel, T., Schuerpf, F. & Carroll, M. C. B cell acquisition of antigen in vivo. Curr. Opin. Immunol. 21, 251–257 (2009).
Bajenoff, M. et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 25, 989–1001 (2006).
Allen, C. D. & Cyster, J. G. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin. Immunol. 20, 14–25 (2008).
Carrasco, Y. R. & Batista, F. D. B cell recognition of membrane-bound antigen: an exquisite way of sensing ligands. Curr. Opin. Immunol. 18, 286–291 (2006).
Nowosad, C. R., Spillane, K. M. & Tolar, P. Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat. Immunol. 17, 870–877 (2016).
Tolar, P., Hanna, J., Krueger, P. D. & Pierce, S. K. The constant region of the membrane immunoglobulin mediates B cell-receptor clustering and signaling in response to membrane antigens. Immunity 30, 44–55 (2009).
Akkaya, M. et al. Toll-like receptor 9 antagonizes antibody affinity maturation. Nat. Immunol. 19, 255–266 (2018). This study reveals that TLR9 stimulation of antigen-bound B cells decreases their ability to internalize, process and present antigen and therefore diminishes B cell–T cell interactions required for the initiation of GC responses; at the same time, it induces extrafollicular differentiation of the B cells in a T cell-independent fashion towards short-lived plasma cells.
Querec, T. et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203, 413–424 (2006).
Querec, T. D. et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10, 116–125 (2009).
Francica, J. R. et al. Analysis of immunoglobulin transcripts and hypermutation following SHIVAD8 infection and protein-plus-adjuvant immunization. Nat. Commun. 6, 6565 (2015).
Green, J. A. et al. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 12, 672–680 (2011).
Schnepp, B. C. & Johnson, P. R. Vector-mediated antibody gene transfer for infectious diseases. Adv. Exp. Med. Biol. 848, 149–167 (2015).
Schnepp, B. C. & Johnson, P. R. Vector-mediated in vivo antibody expression. Microbiol. Spectr. 2, AID-0016–AID-2014 (2014).
Manz, R. A., Lohning, M., Cassese, G., Thiel, A. & Radbruch, A. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 10, 1703–1711 (1998).
Elgueta, R. et al. CCR6-dependent positioning of memory B cells is essential for their ability to mount a recall response to antigen. J. Immunol. 194, 505–513 (2015).
Kim, S. T. et al. Human extrafollicular CD4+ Th cells help memory B cells produce Igs. J. Immunol. 201, 1359–1372 (2018).
Joo, H. M., He, Y. & Sangster, M. Y. Broad dispersion and lung localization of virus-specific memory B cells induced by influenza pneumonia. Proc. Natl Acad. Sci. USA 105, 3485–3490 (2008).
Tolar, P. Cytoskeletal control of B cell responses to antigens. Nat. Rev. Immunol. 17, 621–634 (2017).
Natkanski, E. et al. B cells use mechanical energy to discriminate antigen affinities. Science 340, 1587–1590 (2013).
Luo, W., Weisel, F. & Shlomchik, M. J. B. Cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity 48, 313–326.e15 (2018).
Smith, K. G., Light, A., Nossal, G. J. & Tarlinton, D. M. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 16, 2996–3006 (1997).
Phan, T. G. et al. High affinity germinal center B cells are actively selected into the plasma cell compartment. J. Exp. Med. 203, 2419–2424 (2006).
Ise, W. et al. T follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity 48, 702–715.e4 (2018).
Suan, D. et al. CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity 47, 1142–1153.e44 (2017).
Shinnakasu, R. et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat. Immunol. 17, 861–869 (2016).
Dogan, I. et al. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299 (2009).
Kometani, K. et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39, 136–147 (2013).
Pape, K. A., Taylor, J. J., Maul, R. W., Gearhart, P. J. & Jenkins, M. K. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011). This study shows that owing to their high affinity and ability to be activated in the presence of neutralizing antibodies, isotype-switched IgG-expressing memory B cells prevail during the early phase of immune memory; however, once they disappear due to a shorter life span, IgM + memory B cells take on and lead the later phases of memory response.
Pape, K. A. et al. Naive B cells with high-avidity germline-encoded antigen receptors produce persistent IgM+ and transient IgG+ memory B cells. Immunity 48, 1135–1143.e4 (2018).
Zuccarino-Catania, G. V. et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 15, 631–637 (2014).
Koike, T., Harada, K., Horiuchi, S. & Kitamura, D. The quantity of CD40 signaling determines the differentiation of B cells into functionally distinct memory cell subsets. eLlife 8, e44245 (2019).
Purtha, W. E., Tedder, T. F., Johnson, S., Bhattacharya, D. & Diamond, M. S. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J. Exp. Med. 208, 2599–2606 (2011).
Leach, S. et al. Requirement for memory B cell activation in protection from heterologous influenza virus reinfection. Int. Immunol. 31, 771–779 (2019).
Andrews, S. F. et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl Med. 7, 316ra192 (2015).
Keating, R. et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 14, 1266–1276 (2013).
Burton, B. R. et al. Variant proteins stimulate more IgMGC B-cells revealing a mechanism of cross-reactive recognition by antibody memory. eLlife 7, e26832 (2018).
Portugal, S., Obeng-Adjei, N., Moir, S., Crompton, P. D. & Pierce, S. K. Atypical memory B cells in human chronic infectious diseases: an interim report. Cell Immunol. 321, 18–25 (2017).
Crotty, S. T. Follicular helper cell biology: a decade of discovery and diseases. Immunity 50, 1132–1148 (2019).
Papa, I. & Vinuesa, C. G. Synaptic interactions in germinal centers. Front. Immunol. 9, 1858 (2018).
Acknowledgements
This work was supported by the Intramural Research Program of the US National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks D. Burton and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Akkaya, M., Kwak, K. & Pierce, S.K. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol 20, 229–238 (2020). https://doi.org/10.1038/s41577-019-0244-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-019-0244-2
This article is cited by
-
B cell memory: from generation to reactivation: a multipronged defense wall against pathogens
Cell Death Discovery (2024)
-
Distribution of multi-level B cell subsets in thymoma and thymoma-associated myasthenia gravis
Scientific Reports (2024)
-
MVA-based vaccine candidates encoding the native or prefusion-stabilized SARS-CoV-2 spike reveal differential immunogenicity in humans
npj Vaccines (2024)
-
Nanotechnology of inhalable vaccines for enhancing mucosal immunity
Drug Delivery and Translational Research (2024)
-
Defective mitochondria remodelling in B cells leads to an aged immune response
Nature Communications (2024)
