Article Text

Abstract
Clinical heterogeneity, unpredictable course and flares are characteristics of systemic lupus erythematosus (SLE). Although SLE is—by and large—a systemic disease, occasionally it can be organ-dominant, posing diagnostic challenges. To date, diagnosis of SLE remains clinical with a few cases being negative for serologic tests. Diagnostic criteria are not available and classification criteria are often used for diagnosis, yet with significant caveats. Newer sets of criteria (European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) 2019) enable earlier and more accurate classification of SLE. Several disease endotypes have been recognised over the years. There is increased recognition of milder cases at presentation, but almost half of them progress overtime to more severe disease. Approximately 70% of patients follow a relapsing-remitting course, the remaining divided equally between a prolonged remission and a persistently active disease. Treatment goals include long-term patient survival, prevention of flares and organ damage, and optimisation of health-related quality of life. For organ-threatening or life-threatening SLE, treatment usually includes an initial period of high-intensity immunosuppressive therapy to control disease activity, followed by a longer period of less intensive therapy to consolidate response and prevent relapses. Management of disease-related and treatment-related comorbidities, especially infections and atherosclerosis, is of paramount importance. New disease-modifying conventional and biologic agents—used alone, in combination or sequentially—have improved rates of achieving both short-term and long-term treatment goals, including minimisation of glucocorticoid use.
- lupus erythematosus
- systemic
- lupus nephritis
- therapeutics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
SLE: a challenging disease with a fascinating chronicle
Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disease of variable severity and course, characterised by a tendency for flare (figure 1).1 In SLE, both innate and adaptive immune responses are involved. Interaction of genes with environmental factors leads to numerous immunologic alterations that culminate into persistent immune responses against autologous nucleic acids. Tissue damage—caused by autoantibodies or immune-complex depositions—occurs in kidneys, heart, vessels, central nervous system, skin, lungs, muscles and joints leading to significant morbidity and increased mortality.1

Natural history of SLE and the potential impact of a treat-to-target strategy. The disease starts with a preclinical, asymptomatic phase characterised initially by the appearance of autoantibodies common to all autoimmune diseases and, later, of lupus-specific autoantibodies. Subsequent clinical course is characterised by periods of variable disease activity (measured by SLE disease activity indices), with frequent flares resulting in inflammation-driven irreversible damage. Damage—measured by the SLICC/ACR damage index—increases the morbidity and mortality in SLE. Damage is driven initially by inflammation and later—with progression of the disease—also by therapy. With time, comorbidities such as infections, premature atherosclerosis and malignancies become an important part of the disease burden. Effective therapy targeting low-disease activity or remission has the potential to decrease the frequency and severity of lupus flares and resulting damage. ACR, American College of Rheumatology; SLE, systemic lupus erythematosus.
The chronicle of lupus in the stream of medical history is fascinating.2–4 Hippocrates (460–375 BC) may have first described the disease, calling it herpes esthiomenos (ἕρπης ἐσθιόμενος) or ‘gnawing dermatosis’. Herbernus of Tours applied the term lupus to a skin disease in 916 AD. In 1872, Kaposi subdivided lupus into the discoid and systemic, introducing the concept of systemic disease with a potentially fatal outcome.
Major milestones in the history of SLE include the description of the lupus erythematosus cell; the appreciation of its familial aggregation; the recognition of the lack of a typical disease pattern and the need to consider the overall picture for its diagnosis; and the discovery of the New Zealand Black/New Zealand White F1 lupus mouse model. In 1954, hydralazine-induced lupus was described and in 1982 the ACR classification criteria for SLE were published. During 1964–1990, the treatment of severe SLE with high doses of glucocorticoids and immunosuppressive/cytotoxic drugs was introduced. In 2011, the first biologic therapy for SLE (belimumab, Benlysta) was approved.
In this update, we are discussing evolving concepts in SLE. Of necessity, this is not a comprehensive review. We discuss selected studies—most published within the last 5 years—highlighting their impact on the field and the care of lupus patients. At the same time, through the extensive use of Tables, Figures, Algorithms and Boxes, we provide practical, easy to use information for its management.
Epidemiology and causes
Epidemiology and burden: milder cases in community-based registries but progression over time
SLE has a striking female predominance, with almost 10 women patients for every man affected by the disease. Incidence ranges between 0.3–31.5 cases per 100 000 individuals per year and has increased in the last 40 years, probably due to recognition of milder cases. Adjusted prevalence rates worldwide are approaching or even exceeding 50–100 per 100 000 adults.5 In community-based Caucasian registries, most patients are middle-aged women and approximately 50% of cases are mild at presentation (figure 2A).5 However, a proportion of patients may progress in severity, so that mild, moderate and severe cases are equally split over time to one-third in each category (figure 2B).5 6 Disease severity may vary according to ethnic background and is generally worse in patients of African ancestry and Latin Americans. Health-related quality of life is greatly compromised.7 Annual direct (health care-related) costs are highly related to the severity of the disease and organ(s) involved8 and are estimated to be at least US$3000–12 000 in the USA and €2500–5000 in Europe for patients with moderate to severe disease.8–10
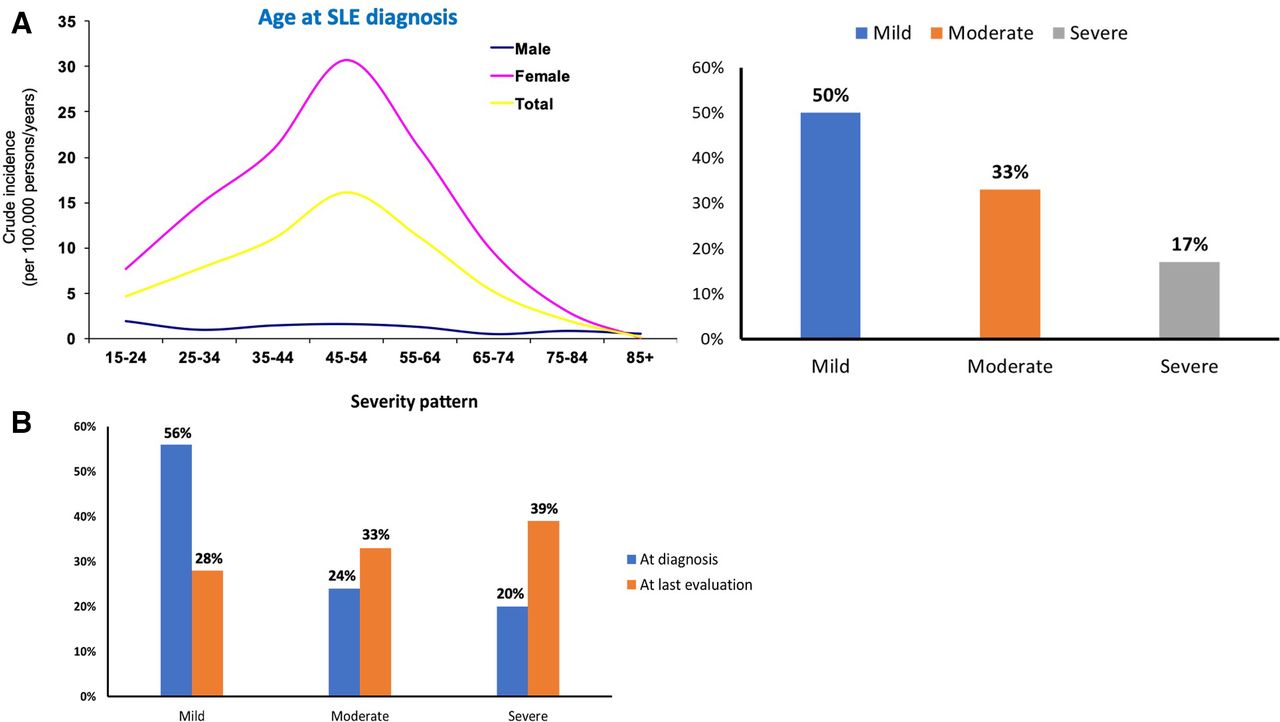
(A) Prevalence and disease severity in SLE. In community-based registries, most patients are middle-aged women and approximately 50% of cases have a mild disease at presentation. In contrast, in tertiary referral centres, most cases have moderate or severe disease. Data from Gergianaki et al.5 (B) Disease progression in SLE. Although most patients with SLE initially present with mild disease, a proportion may progress in severity, so that mild, moderate and severe cases are equally split over time to one-third in each category. Data from Nikolopoulos et al.6 SLE, systemic lupus erythematosus.
Environmental factors, heritability and co-segregation with other autoimmune diseases
Ultraviolet radiation, smoking and drugs are well-established environmental factors linked to SLE pathogenesis.1 At least 118 drugs have been associated with induced lupus, particularly procainamide and hydralazine, while anti-tumour necrosis factor agents (infliximab, adalimumab, etanercept) have been linked to anti-DNA antibody production.11 Among all lupus-related autoantibodies, antiphospholipid (aPL) and anti-DNA antibodies have been associated with smoking.12 13
In general, a polygenic additive model with familial aggregation of SLE cases and also with other autoimmune diseases has been recognised. In a nation-wide study from Taiwan, the relative risks (RRs) for SLE were 315.9 for twins of patients with SLE, 23.7 for siblings, 11.4 for parents, 14.4 for offspring and 4.4 for spouses without genetic similarity.14 The concordance of SLE in monozygotic twins has been estimated to be around 25%.15 16 Genetically determined heritability was calculated at 43.9%, whereas shared (‘familial’) and non-shared environmental factors accounted for 25.8% and 30.3% of SLE susceptibility, respectively. RRs in individuals with a first-degree relative with SLE for various autoimmune diseases vary from 5.87 for primary Sjögren’s syndrome (SS), 5.40 for systemic sclerosis, 2.95 for myasthenia gravis, 2.77 for inflammatory myositis, 2.66 for rheumatoid arthritis (RA), 2.58 for multiple sclerosis, 1.68 for type 1 diabetes mellitus, 1.39 for inflammatory bowel diseases, to 0.86 for vasculitis; these data can provide useful guiding information in counselling families with affected members and provide a basis for understanding the association (or lack of) with other autoimmune diseases. The familial aggregation of primary SS, SLE and RA has been delineated by use of whole-exome sequencing in 31 families with autoimmune rheumatic diseases; rare genetic variations in T-cell receptor signalling pathway seem to be the common denominator for this aggregation.17
All lupus phenotypes: ‘great and small’
Diagnosis of SLE and the confusion with classification versus diagnosis; ‘choosing wisely’ in SLE
Key disease features and their frequency at disease onset and cumulatively can be found in figure 3.18 Diagnosis can be challenging in (1) early stages of the disease, when a limited number of features may be present; (2) antinuclear antibody (ANA)-negative cases or organ-dominant forms and (3) rare disease presentations, which can nonetheless be severe and require prompt treatment. In our experience, non-rheumatologists fail to look consistently for arthritis and to take into consideration features of the disease not present simultaneously. A negative ANA test cannot rule out SLE diagnosis, because up to 20% of patients may be negative (true or false negative) at various stages of the disease, although typically the rate of ANA-negative lupus is much lower.19 Other ‘unwise choices’ include (a) repeating of ANA testing (if once positive), (b) frequent testing of serology in patients with steadily improving or inactive disease and (c) omitting urinalysis from the routine laboratory check. Similar to other chronic diseases, physicians often fail to rule out non-lupus-related causes when trying to explain patient symptoms, with the tendency to attribute them to lupus. Among the many mimics of SLE, viral infections or parasitic infections such as leishmaniasis and lymphoid malignancies need to be considered and excluded.20

Key features and organs involved in SLE. Cumulative frequencies are depicted. Of note, the frequency of nephritis is not as common as previously reported, which may have been the result of referral biases in major lupus centres. Neuropsychiatric disease is an emerging frontier in lupus care. Childhood SLE has higher activity at presentation, is more likely to be severe and to receive more aggressive therapy, as well as accumulate damage. Data from Nikolopoulos et al.18 SLE, systemic lupus erythematosus.
The diagnosis of SLE is clinical, supported by laboratory investigation indicative of immune reactivity or inflammation in various organs. Newer sets of classification criteria21–23 enable the earlier classification of SLE, with the combination of all three sets (ACR-1997, SLICC-2012 and EULAR/ACR-2019) ensuring the capturing of non-overlapping groups of patients (although at the expense of reduced specificity).18 ANA or other immunologic positivity (autoantibodies or hypocomplementemia) are required for classification of SLE according to the SLICC-2012 and EULAR/ACR-2019, but not the ACR-1997 criteria. Fulfilment of the classification criteria is not necessary for the diagnosis for SLE. In patients with early disease, the SLICC and EULAR/ACR are more sensitive than the ACR, while the EULAR/ACR criteria have superior specificity. In spite of this superb performance, some patients with potentially severe disease can still be missed. Modification of the classification criteria may enhance their sensitivity, allowing earlier diagnosis and treatment of more patients with high disease burden (figure 4).24 25

Diagnostic approach to a patient with suspected SLE and the use of classification criteria to aid clinical diagnosis. The diagnosis of SLE is clinical, supported by laboratory abnormalities including serologic assays. Diagnostic criteria are not available for SLE and classification criteria are often used as such, but with several caveats. Among classification criteria, the EULAR/ACR-2019 have the best combination of sensitivity and specificity but require positive ANA as an entry criterion. However, for diagnosis, some patients may be ANA-negative; in such cases, low complement levels and/or positive anti-phospholipid antibodies could be used as an alternative entry criterion in the classification algorithm. For patients who fall short of the classification threshold (ie, EULAR/ACR score <10), inclusion of photosensitivity (defined as in the ACR-1997 criteria) or a combination of immunological and clinical features can still be used for SLE diagnosis. ACR, American College of Rheumatology; ANA,antinuclear antibody; EULAR, European League Against Rheumatism; SLE,systemic lupus erythematosus.
Endotypes and organ dominant lupus
Among the various endotypes, childhood-onset SLE (cSLE), organ-dominant SLE (dermatologic, musculoskeletal—so called ‘rhupus’—, renal, neurological, haematologic), lupus with antiphospholipid syndrome (APS) and SS have received more attention due to differences in prognosis and treatment (online supplemental figure S1). cSLE has higher activity at presentation and is more likely to be severe and to receive more aggressive therapy, as well as accumulate damage. The presence of APS increases the risk of neuropsychiatric SLE (NPSLE), thrombotic and obstetric complications.1 In our experience, up to one-third of patients with apparent primary APS can manifest lupus-like features. Similarly, patients with presumed idiopathic thrombocytopenic purpura, haemolytic anaemia, serositis, APS or SS have increased risk of developing SLE compared with matched controls.26 27
Supplemental material
Supplemental material
Clinical course, activity patterns and adverse prognostic factors
In a large Canadian cohort, approximately 70% of patients with SLE followed a relapsing-remitting course, with the rest 30% divided equally between prolonged remission and persistently active disease (online supplemental figure S2).28 Higher remission rates have been reported from Italy, with 37% of patients achieving prolonged remission; vasculitis, glomerulonephritis and haematological disease were associated with an unremitting disease.29 Remission for at least two consecutive years is associated with halting of damage accrual.30 cSLE, male patients, patients with low complement, positive anti-DNA or aPL antibodies, patients with high interferon (IFN) signature and patients with moderate to high activity indices are more likely to develop severe SLE.1 Such patients should be ideally referred to centres where multidisciplinary care is offered, returning to their physician once a therapeutic plan is in place.
Supplemental material
Metrology and the rationale for measuring activity and damage indices
Due to the multiorgan involvement, there is a need for use of both global and organ-specific, validated disease activity indices to guide therapy and to serve as outcome for clinical trials. Three are the most widely used instruments: (1) SLE Disease Activity Index (SLEDAI); (2) British Isles Lupus Activity Group (BILAG) index and (3) Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA)-SLEDAI Physician Global Assessment (PGA).31 Each index scores general signs and symptoms of disease activity in various organs, with the SLEDAI also scoring lupus serology, such as anti-dsDNA and serum complement levels. The SLEDAI is weighted, while BILAG provides a comprehensive set of definitions for mild, moderate and severe activity in multiple organs and according to the intention-to-treat concept (eg, BILAG A necessitates the use of high-dose glucocorticoids and/or immunosuppressives). PGA should complement objective activity indices, because the latter can miss certain items of disease activity or lack sensitivity to longitudinal changes. In our practice, we use the SLEDAI-2K version of SLEDAI (which allows persistent, rather than new-onset only, activity in alopecia, mucosal ulcers, rash and proteinuria to be scored),32 combined with PGA, and the SELENA-SLEDAI definitions for flares (table 1). A newly proposed SLE Disease Activity Score (SLE-DAS; accessible at http://sle-das.eu/) with more items to include less common—yet severe—manifestations such as myositis, haemolytic anaemia, cardiopulmonary and gastrointestinal manifestations, has improved sensitivity to changes compared with the SLEDAI, while maintaining high specificity and easiness of use.33
Features, caveats and pitfalls of main indices used in SLE: the SLEDAI-2K, the SELENA-SLEDAI Flare Index and the SLICC/ACR Damage Index
In SLE, organ damage assessed by the SLICC/ACR Damage Index (SDI)34 (table 1) is associated with adverse clinical outcomes and death. Although some SDI items are obscurely defined, it currently represents the single, validated and easy-to-use clinical tool to monitor complications or dysfunction across a wide range of organs due to active SLE, administered treatments (especially glucocorticoids) or associated comorbidities. With a maximum score of 46, any increment in the SDI is clinically and prognostically significant, reflecting the burden of the disease.
The use of validated activity and damage indices has been included in the EULAR guidelines for the management of SLE,34 which recommend assessment of at least one activity index at each visit and of SDI once yearly. Free online calculators for both instruments can be found at https://qxmd.com/calculate/calculator_335/sledai-2k and https://qxmd.com/calculate/calculator_336/slicc-acr-damage-index.
Outcome measures and remedying the failure of multiple lupus trials
During the past three decades, late-phase (IIb and III) clinical development programmes involving at least 40 novel agents have failed. While earlier trials from the Mayo Clinic and the United States National Institutes of Health used organ-specific outcome measures (for instance, in nephritis), subsequent trials have employed global outcome measures to capture general SLE activity and response.33 35–37 In the belimumab trials, the SLE Responder Index (SRI) was developed as a composite outcome incorporating a modification of SELENA SLEDAI, BILAG, and a 0-3 Visual Analogue Scale of PGA to determine patient improvement. The BILAG-based Composite Lupus Assessment (BICLA), developed based on data from clinical trials of epratuzumab, requires patients to meet response criteria across three assessment tools, namely SLEDAI, BILAG and PGA. Not unexpectedly, differences in the structure of these two composite indices are reflected on differences in the response rates in recent SLE clinical trials.
A better appreciation of disease heterogeneity and its course; the lack of synchronisation of involvement and timing of response of different end-organs; the differential response of patients of various ancestries and geographic locations; the inclusion of patients with mild disease; the high dose of glucocorticoids and other background medications used; and finally, shortcomings of trial inclusion criteria (such as serology, biomarkers) and endpoints38 have led the community to believe that improved metrics for treatment response are needed and the use of organ-specific endpoints should be reconsidered. These caveats have forced investigators in more recent trials to pay more attention to these issues and modify designs and outcome measures accordingly. Molecular taxonomy and novel biomarkers for diagnosis, monitoring and treatment are available, but need to be further defined. For instance, while 75% of SLE patients have an IFN signature, only 50% of the patients respond to IFN-a inhibitors.36
An attempt has been made to define more rigorous response criteria and disease states, such as remission and lupus low-disease activity state (LLDAS)33 34 39 (online supplemental box 1). LLDAS is a pragmatic and clinically relevant outcome, taking into consideration that (a) remission in SLE is desirable, but not always achievable, and (b) patients who spend more than 50% of their observed time in LLDAS have significantly reduced damage accrual.40 Flare is an emerging trial outcome defined as any increase in disease activity leading to intensification of therapy.
Supplemental material
Management of SLE: winning the war by prevailing in multiple battles
General principles, targets of therapy and recommendations
Management recommendations have been published by EULAR in 2008 and were updated in 2019 based on emerging new data.34 41 Of note, these recommendations represent guidance only, not strict instructions.42 43 Treatment goals include long-term patient survival, prevention of organ damage and optimisation of health-related quality of life. Therapy should aim at remission or at least low disease activity and prevention of flares. All lupus patients should receive hydroxychloroquine, at a dose not exceeding 5 mg/kg real body weight (figure 5). During chronic maintenance treatment, glucocorticoids should be minimised to less than 7.5 mg/day (prednisone equivalent) and, when possible, withdrawn. Appropriate initiation of immunomodulatory agents (methotrexate, azathioprine, mycophenolate) can expedite the tapering/discontinuation of glucocorticoids. In persistently active or flaring disease, add-on belimumab should be considered; rituximab or cyclophosphamide (CY) may be considered in organ-threatening, refractory disease. In the recent update, specific recommendations were also provided for cutaneous, neuropsychiatric, haematological and renal disease. Patients with SLE should be assessed for their aPL antibody status, infectious and cardiovascular disease (CVD) risk profile, and preventative strategies should be adjusted accordingly.

EULAR recommendations for the management of SLΕ drugs, treatment strategy, targets of therapy and adjunct therapy. Determination of severity in SLE is based on (a) the involvement of major organs or organ-threatening disease; (b) concomitant activity from multiple non-major organs; and (c) the need for the use of high doses of glucocorticoids and/or immunosuppressive therapy. aPL, antiphospholipid antibody; AZA, azathioprine; BEL, belimumab; CNI, calcineurin inhibitors; CYC, pulse cyclophosphamide; EULAR, European League Against Rheumatism; GC, glucocorticoids; HCQ, hydroxychloroquine; MMF, mycophenolate mofetil; RTX, rituximab; SLEDAI, SLE Disease Activity Index.
Special considerations
Lupus nephritis
Lupus nephritis (LN) is a major cause for morbidity, increased medical expenses and mortality in SLE.44 The life-long risk for severe nephritis is approximately 20%, although older reports may have overestimated these rates. Younger patients, especially males, those with active serology or with active moderate to severe non-renal lupus, are at higher risk for kidney involvement.44 In reference to histological findings, strong predictors for progression into end-stage renal disease (ESRD) include the presence of extensive interstitial fibrosis/tubular atrophy and crescents45 (online supplemental table S1). In a single-centre study from Milan reviewing cases from 1976 until 2016, risk factors for ESRD were male gender, hypertension, increased baseline creatinine, high histological activity and chronicity indices, and no use of maintenance immunosuppression.46 ESRD-free survival rose from 80% to 90% at 20 years, attributed mainly to earlier diagnostic biopsies and prompt institution of immunosuppressive therapy (online supplemental box 2).46
In the updated 2019 EULAR/European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) recommendations for LN,44 the target of therapy was set as a reduction in proteinuria by ≥25% with stable glomerular filtration rate (GFR; ±10% of baseline value) at first 3 months after treatment initiation; reduction by ≥50% in proteinuria at 6 months; and <0.5–0.7 g/24 hours proteinuria at 12–24 months (all with stable GFR).47
In active proliferative LN, initial (induction) treatment with low-dose intravenous CY (500 mg × 6 biweekly doses) or mycophenolate mofetil (MMF; 2–3 g/day, or mycophenolic acid at equivalent dose), both combined with glucocorticoids (pulses of intravenous methylprednisolone, then oral prednisone 0.3–0.5 mg/kg/day) is recommended. Combination of MMF with calcineurin inhibitors or high-dose CY are alternative regimens for patients with nephrotic-range proteinuria and adverse prognostic factors, respectively. Subsequent long-term maintenance treatment with MMF or azathioprine should follow. The need to minimise patient exposure to glucocorticoids has received more attention; in the updated EULAR/ERA-EDTA recommendations, following pulse IV methylprednisolone, recommended starting dose is 0.3–0.5 mg/day prednisone equivalent, which should be tapered to tapered to ≤7.5 mg/day by 3–6 months. Treatment in children follows the same principles as adult disease. EULAR recommendation-based treatment algorithms for proliferative and membranous LN can be found in online supplemental figures S3 and S4.
Supplemental material
Supplemental material
NPSLE: thrombotic or inflammatory and the problem of attribution
Neuropsychiatric events are diverse and most occur around the diagnosis of SLE.48 Among them, seizures, cerebrovascular events and cognitive dysfunction are the most frequent. The risk of ischaemic stroke is more than twofold compared with the general population, with highest RRs within the first year after SLE diagnosis.49 This presents an opportunity for rheumatologists to screen patients for risk factors and intervene early.42 43 Importantly, approximately 60% of strokes occur in the presence of generalised lupus activity, which has implications for their management (see below). Although the majority of events resolve, they are associated with reduced health-related quality of life and excess mortality.48
Cognitive dysfunction is a significant problem in SLE and often occurs with limited or no structural brain abnormalities on conventional MRI. Using functional MRI in the assessment of cognitive function, Barraclough et al50 showed that patients with SLE have poorer performance on a task of sustained attention and altered brain responses, particularly in default mode network regions and the caudate. The study highlighted that patients with SLE are likely to employ compensatory brain mechanisms to maintain cognitive performance and may score similarly to healthy controls in objective measures of cognition, but may fatigue quicker.
Attribution of neuropsychiatric manifestations to SLE (so-called ‘primary NPSLE’) is complex and requires a comprehensive, multidisciplinary approach to rule out mimics (infections, malignancy, comorbidities and others), by considering: (a) risk (‘favouring’) factors such as type and timing of manifestation, presence of generalised, non-neurological disease activity, abnormal neuroimaging and cerebrospinal fluid analysis, positive aPL antibodies; and (b) confounding factors favouring alternative diagnoses.51 New MRI techniques may help to differentiate primary NPSLE from neuropsychiatric events unrelated to lupus. The former is characterised by hypoperfusion in cerebral white matter that appears normal on conventional MRI; we recently showed that co-registration of MRI with dynamic susceptibility contrast MR-measured blood flow in the brain semioval centre suggests primary NPSLE.52
Immunosuppressive therapy is recommended for NPSLE of presumed inflammatory origin, anticoagulation/antiplatelet therapy for manifestations presumed to be thrombotic or embolic, and their combination if both mechanisms are considered possible.34 A large autopsy study that included both patients with NPSLE (70% of which had cerebrovascular accidents, mostly in the context of generalised lupus activity) showed that microthrombi were found uniquely in NPSLE and were associated with C4d and C5b-9 deposits, suggesting that complement deposition may be a key factor in the interaction between circulating autoantibodies and thromboischemic lesions observed in SLE.53 These indirect data support the EULAR recommendation for a low threshold for immunosuppressive treatment in stroke, especially in the presence of generalised lupus activity and absence of aPL antibodies and atherosclerotic risk factors. An algorithm for the management of NPSLE can be found in online supplemental figure S5.
Supplemental material
Haematological disease and emerging haematologic phenotypes
Autoimmune cytopenias are common in SLE. Haematological manifestations necessitating immunosuppressive treatment in patients with SLE include immune thrombocytopenia (see online supplemental figure S6 for its management) and haemolytic anaemia.34 The presence of thrombocytopenia should prompt examination of the peripheral smear to exclude microangiopathic haemolytic anaemia (MAHA) and thrombotic microangiopathy (TMA). MAHA is non-immune haemolysis resulting from intravascular red blood cell fragmentation that produces schistocytes in the peripheral blood smear. TMA is a diverse syndrome that includes, among others, the classical thrombotic thrombocytopenic purpura (TTP) and is characterised by both MAHA and organ damage due to arteriolar and capillary thrombosis, with characteristic pathologic endothelial and blood vessels wall abnormalities that lead to microvascular thrombosis.54 Not all MAHA is caused by a TMA syndrome, but virtually all TMAs cause MAHA and thrombocytopenia. In rare cases, MAHA may be a manifestation of catastrophic APS.
Supplemental material
Most experts agree that TTP and SLE are distinct clinical syndromes and only rarely coexist. Lupus patients may have reduced levels of the metalloproteinase ADAMTS 13, a classical finding in TTP, which may be due to the presence of autoantibodies against the protein; this may pose difficulties in distinguishing SLE from TTP/TMA and overlapping features, such as severe CNS involvement, may make TTP indistinguishable from SLE exacerbation; in such cases, the use of plasmapheresis or rituximab may be considered. However, in most cases, MAHA in SLE responds to immunosuppressive therapy and does not require plasmapheresis.
Macrophage activation syndrome (MAS) is a rare but potentially fatal complication of SLE, presenting with febrile cytopenia mimicking lupus flares. MAS can coincide or follow the diagnosis of SLE and may relapse in up to 10% of patients.55 High-dose glucocorticoids alone are used as first-line therapy; IV immunoglobulin, CY, rituximab and etoposide are also used, with etoposide and CY-based regimens having the best efficacy.55
Pulmonary hypertension and involvement of the heart
Pulmonary arterial hypertension is an infrequent but serious complication of SLE. Recent data suggest two distinct phenotypes, the vasculopathic with low lupus disease activity (‘pure PAH’) and the so-called ‘vasculitic’ with high lupus disease activity, which may be more responsive to immunosuppressive therapy.56 57 Patients with lupus may also develop pulmonary hypertension via other mechanisms: chronic thromboembolic pulmonary hypertension due to non-resolving occlusion of the pulmonary vasculature or, less frequently, pulmonary hypertension secondary to interstitial lung disease causing hypoxaemia.
Although pericarditis is the most frequent heart manifestation, in SLE valvular disease and, less often, myocarditis may be detected. Both SLE and the presence of aPL increase the risk for valvular heart disease.58 59 Myocarditis is rare yet increasingly recognised in SLE after the advent of heart MRI and use of high-sensitive troponin tests.58 59 Antimalarial-induced cardiomyopathy is a rare, probably under-recognised complication of prolonged antimalarial treatment. It presents as a hypertrophic, restrictive cardiomyopathy with or without conduction abnormalities.60
Women’s health, fertility and pregnancy in SLE
The risk of high-grade cervical dysplasia and cervical cancer is 1.5 times higher in women with SLE.61 Accordingly, human papillomavirus immunisation should be recommended in all SLE women. SLE is impacting on personal relationships and the decision to have children.62 Family planning should be discussed as early as possible after diagnosis. Hormonal contraception and menopause replacement therapy (if there is strong indication) can be used in patients with stable/inactive disease and low risk of thrombosis (figure 6).63–65

Women’s health, fertility and pregnancy in women with SLE. SLE is impacting on personal relationships and family planning should be discussed as early as possible after diagnosis. Most women can have successful pregnancies and measures can be taken to reduce the risks of adverse maternal or foetal outcomes. A pregnancy risk stratification should take into account maternal characteristics, disease characteristics (activity, presence of autoantibodies) and received medications. Low disease activity before and during pregnancy and the use of hydroxychloroquine improve pregnancy outcomes. Underuse of low-dose aspirin use and high prevalence of pre-eclampsia risk factors among pregnant women have been recently reported, pointing to a major gap between practices and current recommendations for pregnant SLE women.SLE,systemic lupus erythematosus.
Most women can have successful pregnancies and measures can be taken to reduce the risks of adverse maternal or foetal outcomes. Risk factors for adverse pregnancy outcomes include active SLE; prior or current active LN; hypertension or proteinuria more than 1 g/day; presence of serological activity or aPL antibodies; previous vascular and pregnancy morbidity; and use of prednisone—a surrogate for active disease. In contrast, there are benefits from the use of hydroxychloroquine and antiplatelets/anticoagulants.66 Increased Bb and sC5b-9—early in pregnancy—are strongly predictive of adverse pregnancy outcomes, supporting the role of activation of the alternative pathway complement.67 Low rates of low-dose aspirin use and high prevalence of pre-eclampsia risk factors among pregnant women in a multinational SLE inception cohort have been recently reported, pointing to a major gap between practices and current recommendations for the care of pregnant SLE women.68
Congenital heart block (CHB) may develop in about 1% of fetuses of anti-Ro/SSA-positive women, including SLE. In a nation-wide healthcare registry, individuals with CHB had significantly increased risk for: (a) cardiovascular comorbidity manifested as cardiomyopathy and/or heart failure and cerebral infarction, (b) a systemic connective tissue disorder and (c) developing any of 15 common autoimmune conditions.69
Comorbidities
Infections
The net risk of infection in SLE is associated with both disease-related and treatment-related factors. Patients should receive vaccinations according to the EULAR recommendations.70 Immunisation against seasonal influenza and pneumococcal infection (both PCV13 and PPSV23) is administered preferably during stable disease. Herpes zoster vaccination with the live vaccine (Zostavax) is available for the general population. In 90 patients with stable SLE not receiving intensive immunosuppression, Zostavax was well-tolerated and provoked an immune response.71 Shingrix, a newer non-live vaccine, is safe and more effective to prevent shingles in the general population, although no studies have been performed in patients with lupus.
Patients with SLE may have a variable net state of immunosuppression, thus infection should be treated when in doubt. An elevated C reactive protein makes a bacterial infection more likely than a disease flare.72 Prompt recognition and treatment of sepsis are essential; validated scores, such as the quick Sepsis-related acute Organ Failure (SOFA) score identifies patients at greater risk for a poor outcome in the emergency room or in hospitalised patients, by scoring three variables (altered mental status, tachypnoea and hypotension).
Cardiovascular disease
SLE is an independent risk factor for CVD, attributed both to traditional and to disease-related risk factors, such as persistent disease activity, LN, presence of aPL and use of glucocorticoids.73 Use of statins should be considered on the basis of lipid levels and the presence of other traditional risk factors. Calculation of the 10-year CVD risk using, for instance, the Systematic Coronary Risk Evaluation (SCORE), is recommended,74 although the actual risk is underestimated in patients with SLE. Maintaining blood pressure below 140/90 mm Hg may reduce vascular events, therefore this should be considered the general target for patients with SLE.75 However, patients with blood pressure >130/80 mm Hg and clinical CVD or a high estimated CVD risk (>10%) should be treated to a target< 130/80 mm Hg.76 77 Moreover, patients with renal disease benefit from lower blood pressure targets, that is, below 120/80 mm Hg and the use of renin-angiotensin-aldosterone system inhibitors.44 In a study from the Taiwan National Health Insurance Research Database, SLE was an independent predictor of in-hospital mortality following percutaneous coronary angioplasty (PCI) and was independently associated with overall mortality, repeat revascularisation and major adverse cardiovascular events. The study demonstrates the inherent risks associated with SLE in patients undergoing PCI and highlights the necessity to improve care and secondary prevention strategies for these high-risk patients.78
Malignancies
Rates of malignancies differ in patients with SLE compared with the general population.79 There is an increased risk of haematological, lung, thyroid, liver, cervical and vulvovaginal, but a decreased risk of breast and prostate cancer. The risk for lymphoma is increased approximately threefold and has been linked to increased activity of multiple inflammatory cytokines, as well as possible viral causes.80
Survival and mortality and the impact of income
In Western countries, the all-cause and cause-specific standardised mortality rates significantly decreased over time, likely reflecting the advances in the management of SLE and certain comorbidities. However, mortality rates are particularly high for patients aged less than 40 years. Results are not as good when looking at the global picture for SLE. After a period of major improvement, survival in SLE has plateaued since the mid-1990s in a review of 125 studies.81 In high-income countries, 5-year survival exceeds 95% in both adults and children. In low-income/middle-income countries, 5-year and 10-year survival was lower among children than adults.81
Recent clinical trials: paving the way for the future
New disease-modifying conventional and biologic agents used alone, in combination or sequentially, have improved rates of achieving treatment goals, including minimisation of glucocorticoid use. More specifically, studies have shown that MMF or enteric-coated mycophenolate sodium is equally effective to CY and superior to azathioprine in studies of patients with general lupus or LN.34 82 Calcineurin inhibitors added to standard-of-care induction therapy for LN (so called ‘multitarget’ therapy) may increase complete renal remission rates and maintain remission. The first regimen tested included tacrolimus in combination with MMF and glucocorticoids, as both induction and maintenance therapy.83 84 The AURA-LN phase 2 study tested the novel calcineurin inhibitor voclosporin for efficacy and safety in active LN. Its addition to MMF and glucocorticoids for induction therapy of active LN resulted in a superior renal response, but higher rates of adverse events including death were observed.85 A subsequent phase 3 study recently confirmed superior efficacy without safety concerns (still in abstract form).86
In reference to biologics, new studies have confirmed earlier data on the efficacy of belimumab. In patients with SLE from North East Asia, belimumab significantly improved disease activity and reduced severe flares while reducing prednisone use. A recent study compared organ damage progression in patients who received belimumab in the BLISS long-term extension study with propensity score-matched patients treated with standard of care from the Toronto lupus cohort. Patients receiving belimumab were 61% less likely to progress to a higher SDI score over any given year compared with patients treated with SoC (HR 0.39).87 In adults with active LN, the Efficacy and Safety of Belimumab in Patients with Active LN (BLISS-LN) study, involving 448 patients, met its primary endpoint, demonstrating that a statistically significant greater number of patients achieved primary efficacy renal response over 2 years when treated with belimumab plus standard therapy compared with placebo (43% vs 32%, OR (95% CI) 1.55 (1.04 to 2.32).88
Anifrolumab, a human monoclonal antibody to type I IFN receptor subunit 1, did not have a significant effect on the SRI (primary endpoint) in the Treatment of Uncontrolled Lupus via the Interferon Pathway (TULIP)-1 phase 3 trial. By contrast, the TULIP-2 phase 3 trial used as its primary endpoint the British Isles Lupus Assessment Group (BILAG)–based Composite Lupus Assessment (BICLA), a secondary endpoint from the TULIP-1 trial. A BICLA response requires (1) reduction in any moderate-to-severe baseline disease activity and no worsening in any of nine organ systems in the BILAG index, (2) no worsening on the SLEDAI, (3) no increase of ≥0.3 points in PGA, (4) no discontinuation of the trial intervention and (5) no use of medications restricted by the protocol. The discrepancy of the results in TULIP-1 and TULIP-2 may be due to differences in the pathophysiology of various organs involved in SLE, differences between the SRI and the BICLA (for instance, SRI counts only complete responses, while BICLA also counts partial responses) and differences in the respective weights of the various organs involved and the serology (BICLA does not take serology into account).37
Open questions, unmet needs emerging and future therapies
SLE continues to be a challenging and disabling disease, but there is now a better understanding of its causes, earlier recognition of its symptoms and signs, and more effective and less toxic drugs. Following the approval of belimumab,89 90 advances in lupus research have led to new clinical trials for investigational drugs, each with a unique mechanism of action (figure 7). These include, but are not limited to, antibodies targeting B-cells or T-cells or their interaction, dendritic cells, IFN and other cytokines, and finally, low-dose IL-2 to boost regulatory T-cell function. Recent successes, such as the baricitinib trial91 and the positive results from the TULIP-2 study of anifrolumab,36 as well as low-dose IL-2,92 provide room for cautious optimism. NPSLE is an emerging frontier for lupus research and care, encompassing a wide spectrum of clinical manifestations and complex pathophysiologic mechanisms that remain poorly understood.93 Treatment of other aspects of SLE, such as skin, neuropsychiatric and haematologic disease, or of symptoms such as fatigue and headache, continues to be problematic. Whether there is a molecular, biological or imaging signature for these endotypes is not clear. To this end, development of organ-specific outcome measures (such as the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI), for cutaneous lupus) may facilitate drug development for different subtypes of the disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pathogenesis and novel therapies in SLE. In SLE, genetic and environmental interactions culminate into aberrant regulation of both innate and adaptive immune responses, with excessive production of IFN-α and autoantibodies. Aberrant lymphocyte activation due either to altered activation threshold and/or defective T-regulatory cell function are key pathogenetic features of SLE. The cells and molecules of the immune system that have been targeted or are in the process of testing for clinical efficacy in SLE are shown in the figure: (1) B-cell (1; 2) plasma cell; (3) B–T-cell co-stimulation; (4) IFNs or their receptors; (5) intracellular kinases; (6) cytokines or their receptors. Combination therapy targeting both innate and adaptive immune responses may be more effective in assuring major, sustained clinical responses in SLE. Figure on the right modified from Klavdianou K, Lazarini A and Fanouriakis A. BioDrugs 2020;34:133–147. APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; BTK, Bruton’s tyrosine kinase; CD, cluster of differentiation; ICOS, inducible T-cell costimulatory; ICOSL, ICOS ligand; IFN, interferon; IL, interleukin; JAK, Janus kinase; PC, plasma cell; pDC, plasmacytoid dendritic cell; SLE, systemiclupus erythematosus; STAT, signal transducer and activator of transcription; TACI, transmembrane activator and CAML interactor.
Acknowledgments
We thank Dr D Vassilopoulos for critical review of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Twitter @none
Contributors DTB conceived and drafted the manuscript. NT contributed to preparation of figures. AF and GB edited the manuscript. All authors read and approved the final form.
Funding Part of this work has been supported by research grants from FOREUM (Foundation for Research in Rheumatology) to DTB and GB and from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation programme (grant agreement no 742390) to DTB.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.