Article Text

Abstract
Objectives To examine the safety and efficacy of rontalizumab, a humanised IgG1 anti-interferon α (anti-IFN-α) monoclonal antibody, in patients with moderate-to-severe systemic lupus erythematosus (SLE).
Methods Patients with active SLE were randomised (2:1) to 750 mg intravenous rontalizumab every 4 weeks or placebo (Part 1), and 300 mg subcutaneous rontalizumab every 2 weeks or placebo (Part 2).
Background Hydroxychloroquine and corticosteroids were allowed. Patients taking immunosuppressants at baseline were required per protocol to discontinue. Efficacy end points included reduction in disease activity by British Isles Lupus Disease Activity Group (BILAG)-2004 (primary), and SLE response index (SRI, secondary) at Week 24. Efficacy was also examined by an exploratory measure of IFN-regulated gene expression (interferon signature metric, ISM).
Results Patients (n=238) received rontalizumab (n=159) or placebo (n=79). At baseline, the mean Safety of Estrogens in Lupus Erythematosus National Assessment version of the SLE Disease Activity Index (SELENA-SLEDAI) score in all cohorts was ∼10, and 75.6% of patients had a high ISM (ISM-High). Efficacy response rates by BILAG and SRI were similar between rontalizumab and placebo groups. However, in the exploratory subgroup of ISM-Low patients, SRI response was higher and steroid use was lower in the rontalizumab-treated patients. There was also a reduction in SELENA-SLEDAI flare index rates (HR 0.61, 0.46 to 0.81, p=0.004) in this subgroup. Adverse events were similar between placebo and rontalizumab groups.
Conclusions The primary and secondary end points of this trial were not met in all patients or in patients with high ISM scores. In an exploratory analysis, rontalizumab treatment was associated with improvements in disease activity, reduced flares and decreased steroid use in patients with SLE with low ISM scores.
Trial registration number NCT00962832.
- Systemic Lupus Erythematosus
- Autoimmune Diseases
- DMARDs (biologic)
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is characterised by activation of the innate and adaptive immune systems and by autoantibody production.1 Although current management of SLE has decreased patient mortality,2 disability from accelerated atherosclerosis,3 and accumulation of tissue damage4 contributes to earlymortality.5 Therefore, newer more targeted treatments with improved safety profiles are still needed.6 ,7
A large body of evidence has implicated interferon α (IFN-α) in SLE.8 ,9 Elevated serum levels of IFN-α in patients with SLE correlate with disease activity and flares in cross-sectional studies.10–15 Therapeutic use of recombinant IFN-α(2a) can lead to a reversible form of drug-induced lupus,16 and genome-wide association studies have identified risk polymorphisms in SLE within IFN-regulated genes.17–19 Microarray of mononuclear cell RNA has shown IFN-inducible gene overexpression in both adult20 ,21 and paediatric22 patients.
Rontalizumab is a human anti-IFN-α monoclonal antibody that neutralises all 12 IFN-α subtypes, preventing signalling through the type I IFN receptor. Rontalizumab does not bind to IFN-β or IFN-ω. Based on acceptable safety and pharmacodynamic effects in a Phase I study in patients with mild SLE,23 a multicentre, randomised, placebo-controlled Phase 2 efficacy and safety trial (ROSE: Rontalizumab in Systemic Lupus Erythematosus) was conducted with rontalizumab.
Methods
Study design
This multicentre, randomised, double-blind, placebo-controlled trial comprised two sequential placebo-controlled substudies (figure 1) (Part 1 and Part 2; 24 weeks each), followed by an open-label safety extension study (Part 3, up to 144 weeks). Part 1 started in September 2009; Part 2 started in August 2010. Patients completing Part 1 or Part 2 were eligible for the open-label safety extension (Part 3). Patients who did not enter Part 3 returned for monthly safety follow-up visits for 48 weeks.

Schema for the two part Phase 2 trial (ROSE) in patients with SLE. FPI, first patient; IV, intravenous; ROSE, Rontalizumab in Systemic Lupus Erythematosus; SC, subcutaneous.
Entry criteria
Eligible patients met ≥ four of the revised American College of Rheumatology classification criteria for SLE,24 and had current or past evidence of an antinuclear antibody titre of ≥1:80. Active disease as defined by the British Isles Lupus Disease Activity Group (BILAG) index25 was required: either severe activity (BILAG A) in ≥ one domain or moderate (BILAG B) disease in ≥ two domains.26 ,27 Key exclusion criteria included active lupus nephritis, unstable neuropsychiatric lupus, severe or unstable antiphospholipid antibody syndrome, current or recent history of systemic bacterial, fungal, viral or parasitic infections, or prior B cell directed therapy (within 12 months). Immunosuppressants were discontinued at randomisation (24–36% of patients). This clinical trial approach has been discussed previously.28 ,29 Patients could receive a short course of oral steroids, 0.25–0.5 mg/kg/day prednisone (or equivalent), during screening followed by a taper to ≤10 mg/day by Week 6 after randomisation. Permitted background treatments included non-steroidal anti-inflammatory drugs, antimalarials, ACE inhibitors, angiotensin receptor-blocking agents, osteoporosis therapies and statins.
Additional rescue treatment for SLE could be provided, but this use constituted a treatment failure. After randomisation, steroid increases exceeding the lowest achieved dose by ≥20 mg for ≥14 days, or any increase exceeding the lowest achieved dose by ≥10 mg for ≥28 days, constituted a treatment failure. At Weeks 20–24, ≥20 mg of prednisone equivalent on any day during this 4-week period, or 10–20 mg/day prednisone equivalent for ≥7 days (cumulative), constituted treatment failure.
Randomisation
After screening, patients in Part 1 were separately randomised 2:1 to receive intravenous rontalizumab 750 mg or placebo intravenous infusion monthly until Week 20. In Part 2, patients were randomised 2:1 to receive subcutaneous rontalizumab 300 mg or a placebo every 2 weeks until Week 22. Randomisation was managed by a central interactive web response system vendor using a permuted blocks randomisation schedule. Patients were stratified by prior use of immunosuppressants within the preceding 12 months and by race/ethnicity.
Efficacy evaluations
Measures of disease activity were the BILAG index, SELENA-SLEDAI, (SLE Disease Activity Index modified for the Safety of Estrogens in Lupus National Assessment trial) and the Physician Global Assessment (PGA). Trial investigators completed extensive training on the use of BILAG and SELENA-SLEDAI scoring, including investigator meeting training, web-based training and verification of training. Enrolment of patients using BILAG criteria was adjudicated by a committee of SLE experts. Evaluations were performed every 4 weeks through Week 36 and every 12 weeks thereafter. Randomised patients with ≥ one postbaseline efficacy assessment were included in all efficacy analyses.
The primary end point was the proportion of patients who at Week 24 achieved reduction of all BILAG A domains present at randomisation to BILAG B or better and all BILAG B domains to BILAG C or better with no new BILAG A or more than one new BILAG B and without being classified as treatment failure due to additional therapies. The key secondary outcome measure was proportion of patients who achieved the SLE response index (SRI)-4 response30 ,31 at Week 24. A prespecified exploratory analysis of the treatment response by these end points in patients stratified by baseline interferon signature metric (ISM) status was conducted.
Other exploratory objectives included percentage of patients with a decreased use of steroids and flare rates defined by the SELENA-SLEDAI Flare Index.32 ,33
Safety assessments
Safety was evaluated by the incidence, nature, severity and drug relatedness of adverse events (AEs), graded according to the National Cancer Institute-Common Toxicity Criteria for Adverse Events (NCI–CTCAE) V.3.0. Lupus flares were captured with the flare outcome measures and not as AEs unless they were serious events.
Diagnostic, pharmacodynamic and other biomarkers
Anti-dsDNA, C3, C4 and antinuclear antibody levels were reported by Covance central laboratories (see online supplementary methods). A bridging-antibody ELISA was validated to detect antibodies to rontalizumab (antitherapeutic antibodies) in human SLE serum.34 Exploratory measures included baseline evaluation of the gene expression ISM, a 3-gene set of IFN-regulated genes used as a surrogate for the global IFN gene expression signature in patients with SLE (Townsend et al,35 submitted). The ISM surrogate (HERC5, EPSTI and CMPK2) for the IFN signature was optimised to detect overall differences in gene expression between identifiable subgroups of patients with lupus. Specific pharmacodynamic evaluations of rontalizumab blockade on IFN-gene expression were assessed by quantitative PCR gene expression changes from seven IFN-regulated genes (IFI27, IFI44, IFIT1, MX1, OAS1, OAS2 and OAS323) at baseline and post dose on Weeks 2, 4, 8, 12, 16, 20, and 24.
Statistical methods
The sample size was selected for adequacy (<15% for the half-width of the 90% CI) to estimate the treatment effect using the primary end point. This proof of concept Phase II study was thus not powered for hypothesis testing, and confidence levels of 90% were employed in this setting. Patients who discontinued the study early were treated as non-responders.
The efficacy-evaluable population comprised patients with postbaseline efficacy assessments. Estimates for treatment effect, corresponding 90% CIs, and exploratory p values were produced using the Cochran-Mantel-Haenszel test, for the responder end points adjusted by the stratification factors used at randomisation while the Cox proportional odds model and the stratified logrank test were employed for time-to-flare analyses. Exact methods (90% CI and Fisher's exact p value) were used for analyses of the ISM-Low subgroup because of the small sample size. Statistical analyses were performed using SAS V.9.2 (The SAS Institute, Cary, North Carolina, USA).
Results
Patient baseline characteristics and disposition
Baseline demographics, SLE disease characteristics and background medications were balanced across treatment groups (table 1). Of 421 patients screened, 238 were randomised and received at least one dose of study drug. Nine (3.8%) patients discontinued the study early (see online supplementary figure S1 and table S5). The intravenous and subcutaneous substudies were conducted sequentially (figure 1), which led to regional differences in racial/ethnic enrolment composition. Two hundred and twenty-nine (96%) patients completed the study through Week 24, including patients who discontinued study drug early but continued in safety follow-up. Post-baseline efficacy assessments were available for 235 patients (efficacy-evaluable population).
Baseline demographics and disease characteristics
ISM as a baseline SLE biomarker
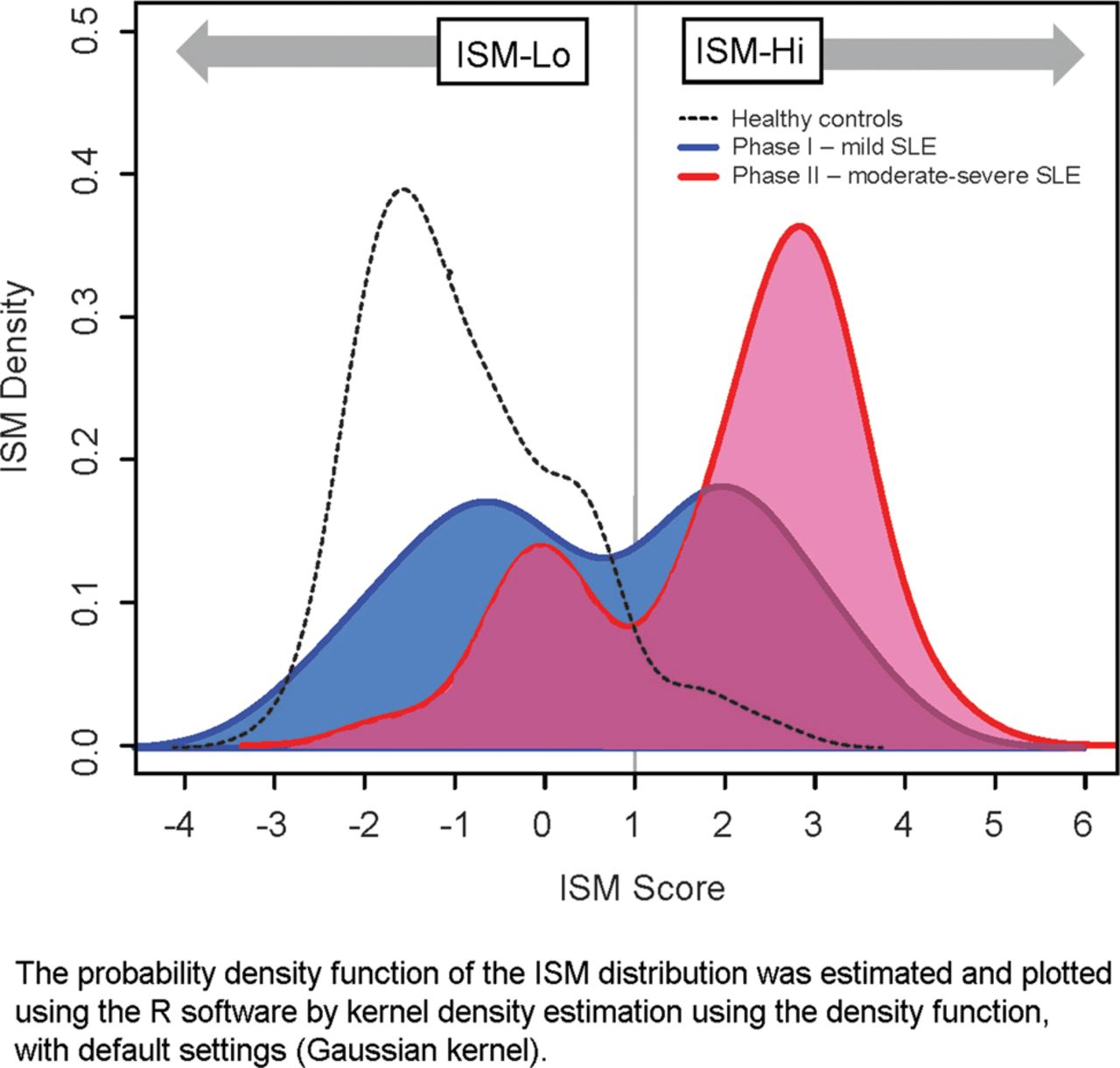
In figure 2, the baseline ISM distribution of patients with SLE in ROSE (n=238) is overlaid with that of patients with milder lupus from a Phase I study of rontalizumab (n=44) and healthy subjects (n=49).18 The probability distribution density function of the ISM scores from patients with SLE in ROSE shows a bimodal distribution. An ISM value of ∼1.0 provides a natural cut-off separating the two subpopulations. This cut-off was used to classify patients with SLE as ISM-High or ISM-Low.

{kind=link}
{kind=link}
Interferon signature metric (ISM) distribution. SLE, systemic lupus erythematosus.
At baseline, 58 patients (24%) were ISM-Low and 180 (76%) were ISM-High. These groups did not differ in baseline disease activity by BILAG scores, SELENA-SLEDAI (see online supplementary table S1) or by disease domain involvement on BILAG. However, ISM-High patients were more likely than ISM-Low patients to have anti-dsDNA autoantibodies (71% vs 35%, respectively), low complement C3 (43% vs 16%) and C4 levels (38% vs 10%) and antiextractable nuclear antigen (anti-ENA) antibodies (73% vs 19%).
Primary efficacy end point: BILAG responder index at Week 24
The primary efficacy end point was the percentage of patients in the treatment versus placebo arms who had step-down improvement in the BILAG score at Week 24 without a new A or >one new B scores, or off-protocol rescue therapies. There was no significant treatment difference (3.5%; p=0.60) in the overall population (pooled intravenous+subcutaneous) or in separate analyses of the intravenous and subcutaneous cohorts. Likewise, no meaningful differences were seen in the ISM low/high subgroups (table 2), whether by pooled or separate study cohort analyses.
Clinical outcomes from ROSE
Secondary efficacy end point: SRI-4 at Week 24
The key secondary efficacy end point was the four-point SRI-4 response at Week 24. Treatment arms did not differ in SRI-4 response for the overall population, the intravenous or subcutaneous cohorts or in pooled analysis (pooled intravenous+subcutaneous treatment difference 5.6%, p=0.41). Response rates in the intravenous study were similar for the placebo (42%) and intravenous rontalizumab (48%) groups, as were rates in the subcutaneous substudy: placebo (50%) and rontalizumab 300 mg (53%). There were no observed treatment effects for the ISM-High subgroup either in the intravenous or subcutaneous studies or in the pooled analysis (table 2). It was hypothesised that patients with the highest IFN gene expression, the ISM-High subpopulation, would be the most likely to benefit from IFN-α blockade. However, the ISM-Low patients appeared to be the most responsive to rontalizumab. The SRI-4 response rates at Week 24 for ISM-Low patients in the intravenous study were 18% and 71% for placebo and intravenous rontalizumab (table 2), respectively. The SRI-4 response rates at Week 24 for ISM-Low patients in Part 2 were 62% and 75% for placebo and rontalizumab (300 mg subcutaneous; table 2), respectively.
Exploratory efficacy end points
An exploratory analysis of the incidence of SELENA-SLEDAI flares showed that over 24 weeks fewer rontalizumab versus placebo patients had flares, with an overall pooled HR estimate of 0.61 (90% CI 0.46 to 0.81) and an associated logrank p value of 0.004 (see online supplementary figure S2). This reduction in flares on active treatment occurred predominantly in the ISM-Low patients and was consistent between the intravenous and the subcutaneous cohorts. In an exploratory analysis of steroid burden, 24% more ISM-Low rontalizumab versus placebo patients maintained an average prednisone dose of ≤10 mg/day between Weeks 8 and 24 (table 3), with consistent results between the intravenous and subcutaneous cohorts.
Exploratory clinical outcomes from ROSE
Safety and tolerability
The incidence of AEs, serious AEs, drug-related AEs, infectious AEs and AEs occurring within 24 h of dosing were comparable between the placebo and rontalizumab groups (see table 4 and online supplementary table S6). No deaths occurred in Part 1 or Part 2 of the ROSE study, but there were two deaths during the open label extension (see online supplementary results) that were attributed to complications of SLE. One malignancy (Stage 0 cervical carcinoma) was reported in a patient in the placebo intravenous group. The most commonly reported AEs were urinary tract infections, upper respiratory tract infection and headaches, with comparable incidence in the treatment groups. Nausea was more common in the rontalizumab group, 12 (8%) patients versus 3 (4%) in the placebo group. No infusion-related or injection-site reactions within 24 h of dosing exceeded Grade 2. Four serious AEs (SAEs) of infection were reported through Week 24: an event of cellulitis in the rontalizumab 300 mg subcutaneous group, a viral infection in the placebo intravenous group, and events of gastroenteritis and pneumonia in the placebo subcutaneous group. SAE due to SLE flare occurred in 6% of rontalizumab patients and 1% of the placebo patients. All the flare SAEs occurred in ISM-High patients, nearly all of whom had very low serum concentrations of rontalizumab (data not shown). Four placebo patients and six rontalizumab patients discontinued study drug due to an AE (5% vs 4%, respectively).
Treatment-emergent AEs in ROSE over 24 weeks*
Discussion
This 24-week study evaluated the effects of rontalizumab in patients with SLE without background immunosuppression. The primary BILAG end point was not met; however, improvement measured by SRI occurred in a higher proportion of rontalizumab-treated patients within a biomarker-defined, immunological subset of patients compared with placebo in an exploratory analysis. In addition, this subgroup also showed decreased use of steroids and flare activity versus placebo. The response subpopulation was first identified in the analysis of Part 1 of ROSE as those patients who were ISM-Low at baseline. These findings were subsequently confirmed in Part 2 of ROSE, which was conducted separately using a subcutaneous dose of rontalizumab. Rontalizumab treatment was generally well tolerated and was not associated with significant safety signals. There was no significant increase in viral or other infectious AEs.
Response to rontalizumab in SLE was expected in the ISM-High rather than the ISM-Low subpopulation. Since approximately 24% of patients in the study were ISM-Low, the response rates observed must be qualified by the relatively low numbers of patients. Nevertheless, there is consistency of responses in this group as measured by clinical improvement, flare reduction, and reduction in steroid use suggesting that IFN-α blockade in this population requires confirmation with larger patient sample sizes.
The response in the ISM-Low subpopulation was not due to obvious differences in baseline symptoms. The ISM-High and ISM-Low patients had comparable baseline disease activity by SELENA-SLEDAI, BILAG and PGA, and had similar organ involvement (primarily musculoskeletal/mucocutaneous). However, ISM-Low patients had, on average, lower anti-dsDNA titres, lower rates of anti-ENA positive serology, and less profound hypocomplementemia (C3, C4) compared with the ISM-High patients (see online supplementary table S2), as has previously been described.36
The ISM-Low patients had higher mean trough concentrations (56.5 μg/mL) of rontalizumab compared with ISM-High patients (39.4 μg/mL), raising the possibility that differing rontalizumab exposure in the two groups may have contributed to the differential outcomes. Comparable attenuation of different IFN-regulated genes, however, occurred in both subpopulations (see online supplementary figure S3).
Rontalizumab treatment was not associated with increased infections or serious infections. In fact, the incidence of all AEs was similar in the rontalizumab and placebo groups. Rontalizumab patients had more low-grade nausea (8%) compared with placebo (4%), but no patients discontinued study drug due to nausea. There was a low rate (3%) of antitherapeutic antibodies to rontalizumab, and they had no apparent impact on the pharmacokinetics or safety during the study. The rate of SLE flares that were SAEs was higher in the rontalizumab (6%) compared with placebo (1%) groups. All of these flare SAEs occurred in ISM-High patients, nearly all of whom had very low serum concentrations of rontalizumab (data not shown). AE rate and deaths were consistent with data from most lupus trials.
Despite the apparent safety of rontalizumab, there are some methodological and design issues in ROSE that limit interpretation of its effects. Use of immunosuppressants such as mycophenolate, azathioprine, cyclophosphamide and methotrexate was restricted (except as rescue therapy) at randomisation and steroid use was controlled during the study. Although this may have eliminated some of the background variables that can complicate interpretation of data, this study was designed to evaluate rontalizumab as a replacement therapy, not, as an add-on therapy. Furthermore, the study was conducted with two different formulations (intravenous, subcutaneous) of rontalizumab and differences in substudy baseline demographics prevented simple pooling of placebo data across the substudies to compare against each rontalizumab arm. Compared with the subcutaneous placebo cohort, the intravenous placebo cohort was younger, had fewer Hispanic patients, a lower incidence of anti-ENA+ autoantibodies (44% vs 63%) and a higher incidence of anti-dsDNA antibodies (66% vs 42%). Larger studies may help to illuminate potential IFN-regulated pathways that may be enriched in populations who were not well represented in our study, since racial differences in autoantibody-IFN relatedness has been observed.37
Accordingly, the placebo response rates in ROSE are divergent between Part 1 and 2. There was a low placebo SRI response of 41% in Part 1 (intravenous) compared with a higher placebo SRI response of 50% in Part 2 (subcutaneous) of ROSE, the difference being greater in the small subset of ISM-Low patients (18% vs 62%) adding to the complexity of the analysis. This might reflect variability and/or differences in patient characteristics due to the low sample size. By comparison, the Phase III studies with belimumab, which used the SRI end point, reported placebo responses of 33.5%38 and 44%.31
Rontalizumab was safe and well tolerated in patients with active extrarenal lupus. This study did not achieve its primary efficacy end point, but exploratory analyses suggested consistent responses at 24 weeks in SRI response,39 flare rates and prednisone reduction within a subpopulation who had entered the trial with a low IFN signature. The corresponding lack of response in ISM-High patients could be due to inadequacy of dose or complex multipathway disease in that subpopulation.
Acknowledgments
The authors thank all of the patients and the investigators who participated in this study. Writing assistance was provided by Genentech.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Correction notice This article has been corrected since it was published Online First.
Contributors All authors contributed to data collection, statistical analysis and writing. All authors have taken due care to ensure the integrity of the work and the final manuscript has been seen and approved by all of them.
Funding Genentech.
Competing interests KCK: Consultant for Genentech; JTM: Consultant for Genentech; RM: Employee and shareholder of Roche; JMB; Employee and shareholder of Roche; MT: Employee and shareholder of Roche; XW: Employee and shareholder of Roche; WPK: Employee and shareholder of Roche.
Patient consent Obtained.
Ethics approval The relevant Institutional Review Boards/Independent Ethics Committees and conducted in accordance with the ethical principles of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement At Roche, we believe that transparency is critical to a business environment that is both productive and responsible. Clinical trial results from Roche sponsored studies have previously been reported on Roche-trials.com and ClinicalTrials.gov, as well as published in journals and at congresses. The expansion of the Roche Data Sharing Policy reflects a commitment by Roche to increasing transparency and sharing of clinical trial information. In developing this policy, we have taken a thoughtful approach that strikes a balance between our global corporate commitment to sharing data, while safeguarding patient confidentiality, and the regulatory process. Universal data sharing is good for scientific advancement and increasing innovation. We are committed to, and enthusiastic about, the promise this offers science and society and the benefits greater openness could ultimately deliver to patients. The Roche Data Sharing Policy is a global policy for both Roche and Genentech on the sharing of clinical trials data. This policy provides the opportunity to request and receive global clinical study reports (CSRs) and other summary reports. In addition, researchers may obtain access to analysable patient-level data from our clinical trials after their requests have been reviewed and approved by an independent panel of experts. Access will be approved by this independent panel on the basis of scientific merit. In both cases, data will be anonymised to respect the privacy of patients participating in our trials in accordance with relevant laws and regulations. Requests for CSRs and other summary reports, as well as analysable patient-level data can be made on this website. Links to study results registries are also provided here.