Article Text

Abstract
Background Intravenous belimumab plus standard of care (SoC) is approved in the USA and Europe for treatment of active, autoantibody-positive systemic lupus erythematosus (SLE).
Methods This phase III, multicentre, randomised, double-blind, placebo-controlled study (BEL113750; NCT01345253) was conducted in 49 centres across China, Japan and South Korea (May 2011–September 2015). Patients with SLE were randomised 2:1 to intravenous belimumab 10 mg/kg or placebo, plus SoC, every 4 weeks until Week 48. The primary endpoint was the SLE Responder Index (SRI) 4 response rate at Week 52. Secondary endpoints were the percentage of patients with ≥4 point reduction in Safety of Oestrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI), SRI7, time to first severe flare and number of days prednisone (or equivalent) dose ≤7.5 mg/day and/or reduced by 50% from baseline. Safety was assessed.
Results The modified intent-to-treat population included 677 patients (belimumab n=451, placebo n=226). At Week 52, the SRI4 response rate was higher with belimumab versus placebo (53.8% vs 40.1%; OR: 1.99 (95% CI: 1.40, 2.82; P=0.0001)). The percentages of patients with a ≥4 point reduction in SELENA-SLEDAI and an SRI7 response were significantly greater for belimumab versus placebo. Patients in the belimumab group had a 50% lower risk of experiencing a severe flare than those receiving placebo (P=0.0004). In patients with baseline prednisone dose >7.5 mg/day, there was a significant reduction in steroid use favouring belimumab (P=0.0228). The incidence of adverse events was similar between groups.
Conclusions In patients with SLE from North East Asia, belimumab significantly improved disease activity, while reducing prednisone use, with no new safety issues.
- Systemic Lupus Erythematosus
- Treatment
- Disease Activity
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a chronic, remitting autoimmune disease resulting in morbidity, mortality and poor quality of life.1–3 Patients with SLE are susceptible to organ damage accrual caused by both active disease and medication toxicities.4 5
Conventional SLE therapies include corticosteroids, antimalarials and non-steroidal anti-inflammatory drugs (NSAIDs) for those without major organ involvement, with immunosuppressive agents prescribed in severe refractory disease.6 For those with high disease activity, there is little evidence for substantial benefits with currently available treatments.6 Moreover, corticosteroid use is associated with significant side effects, particularly in the long term.7 While prognosis of SLE has improved substantially over the last 40 years, there remains an unmet need for an effective treatment with an acceptable safety profile, particularly in those with high disease activity, with corticosteroid reduction remaining an important treatment goal in these patients.8
SLE is associated with elevated levels of B lymphocyte stimulator (BLyS) protein, which enhances B cell proliferation9 and Ig secretion,10 and is critical to B cell survival.11 12 Belimumab is a human, IgG1λ monoclonal antibody that binds soluble human BLyS and inhibits its activity.13–15 Intravenous belimumab 10 mg/kg is approved in the USA and Europe to treat adults with active, autoantibody-positive SLE who are receiving standard of care (SoC).14 15 The safety and efficacy profile of belimumab in this patient group was demonstrated in three phase III studies: Study of Belimumab in Subjects with SLE (BLISS)-52,16 conducted in South America, Asia-Pacific and Eastern Europe; BLISS-76,17 conducted primarily in North America and Europe and Study of Belimumab in Subjects with SLE-Subcutaneous (BLISS-SC), conducted in North, Central and South America, Eastern and Western Europe, Australia and Asia.18 Two open-label extension studies (BEL11223319 and BEL11223420) assessed long-term safety and efficacy in patients who completed BLISS-76 and BLISS-52. In addition to significant improvements in disease activity, the results of these studies suggested that belimumab may have a corticosteroid-sparing effect.16–18
The prevalence of SLE in Mainland China and Hong Kong ranges from 37.7/100 000 to 58.8/100 000,21 22 with some figures ranging as high as 40–100 cases per 100 000 persons and >100/100 000 in women.23 24 In South Korea, SLE prevalence was 26.5/100 000 in 2010.25 The Chinese SLE Treatment and Research Group developed an online registry in 2009 to depict major clinical characteristics of SLE in the Chinese population and compared these against similar observational cohorts in the USA and Europe. The results of the observed clinical manifestations of 2104 patients showed some important differences, most notably an increase in haematological disorders in the Chinese population compared with European cohorts, an increase in kidney disease diagnosis among Chinese patients and fewer neurological manifestations compared with both European and US cohorts.23 Further studies are needed to investigate the epidemiology of SLE within the North East Asian population and the potential genetic and environmental contributory factors.
As numbers of patients from North East Asia were low in previous studies of belimumab, the aim of the present study was to evaluate the efficacy and safety of belimumab as add-on therapy to SoC in patients with autoantibody-positive SLE in North East Asia.
Methods
Study design
This was a phase III, multicentre, randomised, double-blind, placebo-controlled study (BEL113750; NCT01345253) conducted in 49 centres across China, Japan and South Korea between May 2011 and September 2015. Patients completed a screening period of up to 5 weeks, following which they were randomised (2:1) to receive belimumab 10 mg/kg intravenously or placebo, in addition to SoC, on Days 0, 14 and 28 and then every 28 days up to Week 48 (figure 1A). An open-label extension period is ongoing in China.

(A) Study design; (B) Consolidated Standards of Reporting Trials (CONSORT) flow diagram. aFollow-up period for those not entering the open-label period; blast dose for those not entering the open-label period.
Randomisation schedules were created by GlaxoSmithKline (GSK) using validated software (RandAll). The block size was three and the randomisation was stratified by screening Safety of Oestrogens in Lupus Erythematosus National Assessment-SLE Disease Activity Index (SELENA-SLEDAI; ≤9 vs ≥10), complement (low C3 and/or C4 vs no low C3 or C4) and country of origin. GSK’s proprietary interactive voice response system Registration and Medication Ordering System was used for this study. Staff members preparing belimumab and placebo formulations, which were identical in appearance and labelled in a double-blind manner, were not involved in other study activities. A protocol summary is available in the GSK Clinical Study Register (https://www.gsk-clinicalstudyregister.com/).
Patients
Enrolled patients were ≥18 years of age with a clinical diagnosis of SLE according to American College of Rheumatology classification criteria26 and clinically active disease, defined as a SELENA-SLEDAI score ≥8 at screening. Patients also had to have a positive antinuclear antibody test result (either from two independent time points within the study screening period or one positive historical test result and one positive test result during the screening period) and be receiving a stable SLE treatment regimen (corticosteroids (all doses reported as prednisone or equivalent), antimalarials, NSAIDS or any other immunosuppressive or immunomodulatory therapy) for ≥30 days prior to baseline (table 1).
Baseline patient characteristics
Patients with severe lupus kidney disease or active nephritis requiring acute therapy within 90 days prior to baseline or central nervous system (CNS) lupus requiring therapeutic intervention within 60 days prior to baseline and those requiring new SLE medications other than corticosteroids within 60 days prior to baseline were excluded, as were those who had received B cell-targeted therapy at any time. All patients provided written informed consent. Key inclusion and exclusion criteria can be found in the online supplementary material.
Supplementary file 1
Assessments
The primary endpoint was the SLE Responder Index 4 (SRI4) response rate at Week 52 (defined as a ≥4 point reduction from baseline in SELENA-SLEDAI score, no worsening (<0.3 point increase from baseline) in Physician’s Global Assessment (PGA) and no new British Isles Lupus Assessment Group (BILAG) A organ domain score or two new BILAG B organ domain scores vs baseline).27 SRI4 response over time was also analysed.
Secondary endpoints were the percentage of patients with a ≥4 point reduction from baseline in SELENA-SLEDAI score at Week 52, the percentage of patients with an SRI7 response (≥7 point reduction from baseline in SELENA-SLEDAI score, no worsening (<0.3 point increase from baseline) in PGA and no new BILAG A organ domain score or two new BILAG B organ domain scores vs baseline) at Week 52, time to first severe flare (according to the modified SLE Flare Index (SFI)28–30) over 52 weeks and number of days of daily prednisone dose (or equivalent) ≤7.5 mg/day and/or reduced by 50% from baseline over 52 weeks in patients with baseline dose >7.5 mg/day.
Additional prednisone (or equivalent) assessments included the cumulative prednisone dose over 52 weeks and the percentage of patients whose average prednisone dose was reduced by ≥25% from baseline to ≤7.5 mg/day (or equivalent) during Weeks 40–52.
Other endpoints included duration of SRI4 response at 52 weeks and BILAG improvement by organ domain in patients with an A or B domain score at baseline.
Safety variables included: adverse events (AEs), serious AEs (SAEs) and AEs of special interest (AESIs) (malignant neoplasms, post-infusion systemic reactions, all infections, depression/suicide/self-injury and deaths) throughout the study period; Columbia-Suicide Severity Rating Scale (C-SSRS) at every visit; clinically significant changes in haematological or clinical chemistry parameters or urinalysis and immunogenicity. Physical examinations and measurement of vital signs were also carried out.
Statistical analysis
The target sample size was 702 patients (belimumab n=468; placebo n=234) to provide 85% power to detect a 12% treatment difference on the primary SRI4 endpoint (38.6% vs 50.6%) assuming a type I error of 5%.
Safety endpoints were assessed in the safety population, which included all randomised patients who received ≥1 dose of study drug. Efficacy endpoints were assessed in the modified intent-to-treat (mITT) population, which comprised the safety population, with the exclusion of 28 patients from one site (due to compliance issues). For the primary endpoint, sensitivity analyses were carried out in: the completers population, which included all patients in the mITT population who completed all 52 weeks of the planned treatment period; the per-protocol population, which included all patients in the mITT population who completed the study without a protocol deviation and the medications population, which included all patients in the mITT population except those with post-baseline increases in usage of the traditional Chinese medications glucosides of paeony or tripterygium.
Significance tests were performed at a two-sided 5% alpha level. A step-down sequential testing procedure was used to control the overall type I error rate for the testing of the primary and key secondary efficacy endpoints, whereby statistical significance of the primary endpoint led to testing of the secondary disease activity endpoints (percentage of patients with a ≥4 point reduction from baseline in SELENA-SLEDAI score at Week 52 and percentage of SRI7 responders at Week 52), and statistical significance of the secondary disease activity endpoints led to testing of the secondary disease management endpoints (number of days of daily prednisone dose ≤7.5 mg/day (or equivalent) and/or reduced by 50% from baseline over 52 weeks and time to first severe SFI flare over 52 weeks).
The primary endpoint and the secondary disease activity endpoints were analysed using a logistic regression model, with independent variables treatment group, country, baseline SELENA-SLEDAI score and complement levels. The secondary endpoint number of days of daily prednisone dose ≤7.5 mg/day (or equivalent) and/or reduced by 50% from baseline over 52 weeks was analysed using a rank analysis of covariance (ANCOVA) model with independent variables treatment group, baseline prednisone level, country, baseline SELENA-SLEDAI score and complement levels. Data are displayed in a histogram produced post hoc. Time to severe SFI flare over 52 weeks was analysed using a Cox proportional hazards model, adjusting for country, baseline SELENA-SLEDAI score and complement levels.
For the primary endpoint, any patients who dropped out prior to Week 52 and had no double-blind assessment within 28 days of Day 364 and/or used a prohibited medication or a non-allowed dose of restricted medication were considered treatment failures and counted as non-responders in the analysis. Last observation carried forward principles were applied to address other components of missing data. For cumulative prednisone dose, missing data were imputed using the 28-day average prior to last visit.
Results
Study population and patient disposition
Patients were recruited between May 2011 and July 2014, and follow-up for the double-blind analysis continued until September 2015. Of 1116 patients screened, 709 (63.5%) were enrolled and 407 (36.5%) failed screening. The main reasons for screening failure were not meeting inclusion and exclusion criteria (365/407, 89.7%), withdrawal by the patient (29/407, 7.1%) and investigator decision (11/407, 2.7%) (figure 1B). In total, 707 of 709 patients were randomised to receive placebo (n=236) or belimumab (n=471); two patients discontinued and were never randomised. Two further patients were identified as ineligible for inclusion after randomisation and did not receive the first dose of the study drug. Thus, the safety population comprised 705 patients (placebo n=235; belimumab n=470) and the mITT population comprised 677 patients (placebo n=226; belimumab n=451). A total of 567/705 (80.4%) patients in the safety population completed the 52-week study; 56 (23.8%) patients in the placebo group and 82 (17.4%) patients in the belimumab group withdrew prior to study completion (figure 1B). Demographic and baseline disease characteristics were similar between treatment groups (table 1). Most patients were being treated with concomitant steroid medication (placebo n=223 (98.7%); belimumab n=443 (98.2%)). The use of antimalarial concomitant medication was higher in the China cohort (77.0%) compared with the non-China (Korea and Japan) cohort (49.4%). The use of other immunosuppressive/immunomodulatory agents, however, was higher in the non-China cohort (73.1%) compared with the China cohort (62.1%) (see online supplementary table 1).
Efficacy
Significant improvements in favour of belimumab were seen for the primary endpoint, with more patients in the belimumab group achieving an SRI4 response at Week 52 compared with placebo (53.8% (240/446) vs 40.1% (87/217); OR 1.99 (95% CI: 1.40, 2.82; P<0.0001)) (figure 2A). The results of all sensitivity analyses were consistent with the primary analysis (data not shown). Significant differences between the treatment groups were seen from Week 12 and maintained until Week 52. Analysis of the individual components of the primary endpoint demonstrated significant improvements in favour of belimumab for all three parameters (PGA and BILAG data not shown; SELENA-SLEDAI is described below).
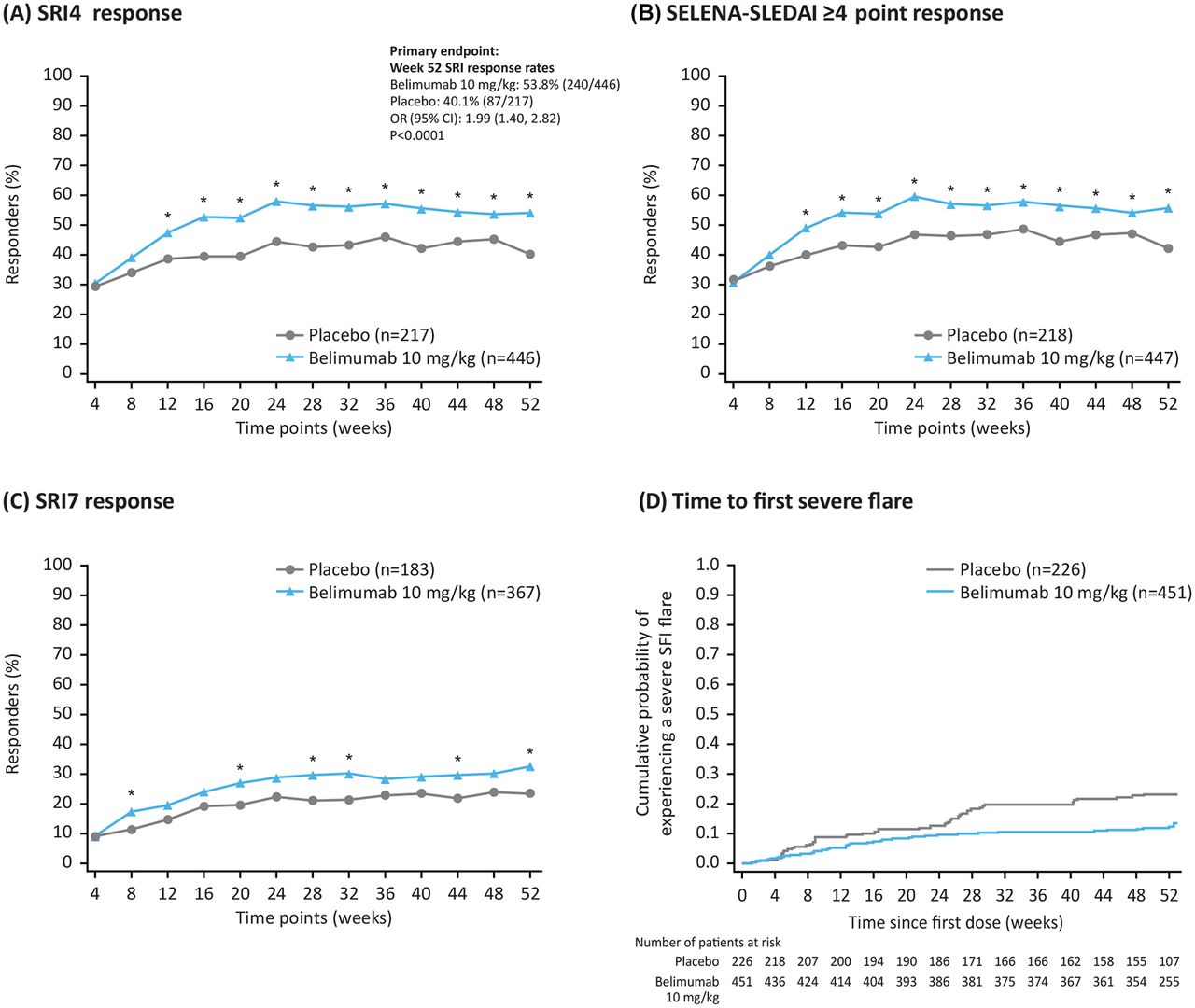
Efficacy endpoints, by visit (modified intent-to-treat population). (A) SRI4 response; (B) SELENA-SLEDAI ≥4 point reduction; (C) SRI7 response and (D) time to first severe flare. *P<0.05 (logistic regression model for belimumab vs placebo with independent variables treatment group, country, baseline SELENA-SLEDAI score (≤9 vs ≥10) and baseline complement levels (low C3 and/or C4 vs no low C3 or C4)). C3/C4, complement 3/complement 4; SELENA-SLEDAI, Safety of Oestrogen in Lupus Erythematosus National Assessment-SLE Disease Activity Index; SFI, SLE Flare Index; SRI, SLE Responder Index.
Significant improvements were also seen for all four key secondary endpoints. A significantly greater proportion of patients in the belimumab group achieved a ≥4 point reduction from baseline in SELENA-SLEDAI score at Week 52 compared with placebo (55.7% (249/447) vs 42.2% (92/218); OR 2.00 (95% CI: 1.41, 2.83; P=0.0001)) (figure 2B). Overall, more patients receiving belimumab achieved an SRI7 response at Week 52 compared with those receiving placebo (32.4% (119/367) vs 23.5% (43/183); OR 1.76 (95% CI: 1.13, 2.74; P=0.0116)) (figure 2C). A lower percentage of patients in the belimumab group experienced a severe flare compared with placebo (12.0% (54/451) vs 22.1% (50/226)), equating to a 50% lower risk in those receiving belimumab (HR 0.50 (95% CI: 0.34, 0.73; P=0.0004)) (figure 2D). In patients with a baseline prednisone dose of >7.5 mg/day, there was a significant reduction in steroid use favouring belimumab (rank ANCOVA, P=0.0288). Although the median number of days that prednisone equivalent dose was ≤7.5 mg/day and/or reduced by 50% was zero in both groups, the 75th centile was larger for belimumab compared with placebo (213.5 vs 172.0 days), reflecting the longer durations of reduced steroid use seen for patients in the belimumab group compared with placebo (figure 3).

{kind=link}
{kind=link}
{kind=link}
Histogram showing the number of days of daily prednisone dose ≤7.5 mg/day (or equivalent) and/or reduced by 50% from baseline among patients with baseline daily prednisone dose >7.5 mg/day (mITT population).aHistogram produced post hoc; the number of days indicates the midpoint of the category, that is, Day 30 represents Days 15–45. mITT, modified intent-to-treat.
Across the population, cumulative prednisone dose (or equivalent) over 52 weeks was significantly lower in the belimumab group versus placebo (P=0.0005; table 2). Compared with the placebo group, more patients in the belimumab group had a dose reduction of ≥25% to ≤7.5 mg/day during Weeks 40–52 (10.9% vs 15.6%; OR 1.68 (95% CI: 0.95, 2.96; P=0.0721)).
Prednisone* dose over 52 weeks (mITT population)
The duration of Week 52 SRI response was summarised by monthly intervals (≥1–≥11 months). A greater proportion of patients in the belimumab group compared with placebo had an SRI response through Week 52 for each monthly duration interval (see online supplementary table 2).
Among patients with an A or B BILAG domain score at baseline, BILAG improvement at Week 52 in favour of belimumab was seen in the haematology (P=0.0445), mucocutaneous (P=0.0250), musculoskeletal (P=0.0078) and general (P=0.0069) organ domains only; however, it is important to note that patient numbers were small (see online supplementary table 3).
Safety
The overall incidence of AEs was similar between the placebo and belimumab groups (75.7% vs 74.9%) (table 3) and most AEs were mild or moderate in severity. The most common AE reported in the placebo and belimumab groups was upper respiratory tract infection (16.6% vs 13.8%, respectively). The incidence of SAEs was higher for placebo (18.3%) than belimumab (12.3%), with similar rates of infectious SAEs reported in both groups (5.5% vs 5.3%, respectively).
Summary of treatment-emergent AEs (safety population)
Incidences of AESIs were similar between the placebo and belimumab groups (table 3). There were no completed suicides and no suicide attempts within the belimumab group; there was one suicide attempt in the placebo group and one case of suicide ideation in the belimumab group. According to the C-SSRS assessment, of patients with no C-SSRS pretreatment suicidal ideation or behaviour, five reported on-treatment suicidal ideation (placebo 2/231 (0.9%); belimumab 3/467 (0.6%)); this included the aforementioned single cases of suicidal behaviour/attempt (placebo group) and severe suicidal ideation (belimumab group). A solid tumour malignancy (cervical carcinoma) was reported in one patient receiving belimumab. One patient in the placebo group experienced fatal respiratory failure after developing cold symptoms/productive cough/dyspnoea; the incident was considered by the investigator to be unrelated to study treatment and no autopsy was performed.
A higher than expected screen failure rate in China was noted for positive hepatitis B virus (HBV) core antibody, and eligibility criteria were subsequently changed in a China-specific amendment so that those with negative hepatitis B surface antigen, but positive HBV core antibody (anti-HBc) and negative HBV DNA by PCR, became eligible for enrolment. As a result of this amendment, a total of 78 patients, representing 16.6% of the safety population (n=470), were randomised into the belimumab treatment arm with positive HBV core antibody and no detectable HBV DNA (HBV DNA by PCR negative). No patients in this study experienced a reactivation of hepatitis B.
There were no clinically meaningful differences between treatment groups in haematology, clinical chemistry or liver function parameters. One patient in the belimumab group had a Grade 3 low IgG value that was a shift from a baseline grade of 0; this patient did not experience an infection within 30 days of the date of the Grade 3 IgG value. The total rate of immunogenic response (a positive anti-belimumab antibody result) was low (placebo 14/235 (6.0%); belimumab 1/470 (0.2%)).
Discussion
This double-blind, randomised, placebo-controlled clinical study assessed the efficacy and safety of intravenous belimumab 10 mg/kg as add-on therapy to SoC in patients with active, autoantibody-positive SLE from North East Asia. Belimumab treatment produced early and sustained improvements in disease activity, while reducing prednisone dose, with a safety profile consistent with previous studies.16–19
The study met its primary endpoint, with a higher proportion of patients treated with belimumab achieving an SRI4 response at Week 52 compared with placebo. Significant differences in favour of belimumab were seen as early as Week 12. Furthermore, significantly higher proportions of patients in the belimumab group achieved each of the three individual response components that made up the composite primary endpoint, including a ≥4 point reduction in SELENA-SLEDAI score (a secondary endpoint), and all sensitivity analyses of the primary endpoint were statistically significant for belimumab at Week 52. These results are consistent with those from the BLISS-52 and BLISS-76 trials of intravenous belimumab, and the BLISS-SC trial of subcutaneous belimumab, performed across several worldwide locations.16–18 Further efficacy benefits demonstrated with belimumab in the present study, compared with placebo, included a significantly higher proportion of patients achieving an SRI7 response at Week 52. The risk of a severe flare was also reduced in the belimumab group, compared with placebo, consistent with results from BLISS-52,16 BLISS-7617 and BLISS-SC.18 Interestingly, when severe flares occurred in the belimumab group, they occurred early in the study, possibly before the full belimumab effect was observed.
As mentioned previously, reducing corticosteroid dose is a key treatment goal in SLE.8 This study builds on findings from previous studies, which suggest that belimumab has a corticosteroid-sparing effect.16–18 In the present study, cumulative prednisone dose over the 52-week treatment period was significantly lower in patients receiving belimumab, compared with those receiving placebo, and dose reductions to ≤7.5 mg/day during the last 12 weeks of the study occurred more frequently in the belimumab group. Our findings are in line with a recent post hoc analysis of the pooled BLISS-52 and BLISS-76 trials, which demonstrated a significantly smaller increase in cumulative corticosteroid dose over 1 year in patients receiving belimumab, compared with those receiving placebo, along with more patients with reductions in oral corticosteroid dose, and fewer patients with increases in oral corticosteroid dose.31
It should be noted that this study was not powered to compare risks against the background of different SoC treatments. As expected, antimalarial use was higher in the China cohort compared with the non-China cohort; the antimalarial hydroxychloroquine was not available in Japan at the time of the study. Moreover, the study excluded some patient populations, including paediatric patients and those with severe active lupus nephritis or CNS lupus, therefore belimumab could not be evaluated in these patient subsets. Furthermore, rare safety signals cannot be ruled out until a much larger number of patients have been treated with belimumab for longer than 52 weeks. Despite these limitations, the study provides valuable information about the effect of belimumab treatment in a North East Asian population of patients with active, autoantibody-positive SLE.
This fourth pivotal belimumab trial in SLE achieved statistical significance for the primary endpoint and all four prespecified key secondary endpoints, including reduction in prednisone use. The reduction in prednisone use in patients on high-dose prednisone (>7.5 mg/day) and the longer duration of reduced use observed within the belimumab treatment group provide encouraging results for belimumab as an add-on to current SoC. Overall, efficacy and safety results were consistent with intravenous and subcutaneous belimumab data until now, with no new safety issues identified and no suicides or suicide attempts in the belimumab group. This study provides evidence that supports the use of intravenous belimumab 10 mg/kg, in addition to SoC, as a viable treatment option for patients with SLE in North East Asia who present with disease activity despite SoC treatment.
Acknowledgments
Medical writing support was provided by Nicole Cash, MRes, PhD, and Louisa Pettinger, PhD, Fishawack Indicia Ltd, UK, and was funded by GlaxoSmithKline (GSK).
References
Footnotes
Handling editor Tore K Kvien
Contributors DB and DAR contributed to the study design, data analyses and interpretation. FZ, S-CB, YT and JZ contributed to the acquisition of data. MC, SE and DG contributed to data analyses and interpretation. All authors reviewed the manuscript and approved the final version for submission.
Funding This study was funded by GSK.
Competing interests DB, MC, SE, DG and DAR are employees of GSK and hold shares in the company. YT receives consulting fees, speaking fees and/or honoraria from AbbVie, Chugai, Daiichi-Sankyo, Bristol-Myers Squibb, Mitsubishi-Tanabe, Astellas, Takeda, Pfizer, Asahi-Kasei, YL Biologics, Sanofi, Janssen, Eli Lilly and GSK and has received research grants from Mitsubishi-Tanabe, Takeda, Daiichi-Sankyo, Chugai, Bristol-Myers Squibb, MSD, Astellas, AbbVie and Eisai.
Patient consent Obtained.
Ethics approval The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, International Council for Harmonisation on Good Clinical Practice and the applicable country-specific regulatory requirements.
Provenance and peer review Not commissioned; externally peer reviewed.
Presented at Data from this study have previously been presented at the American College of Rheumatology Annual Scientific meeting (ACR2016), the 12th International Congress on Systemic Lupus Erythematosus (LUPUS 2017) and the Japan College of Rheumatology 61st Annual Scientific Meeting (JCR 2017).