Article Text

Abstract
Objective To assess the efficacy and safety of the type I interferon receptor antibody, anifrolumab, in patients with active, biopsy-proven, Class III/IV lupus nephritis.
Methods This phase II double-blinded study randomised 147 patients (1:1:1) to receive monthly intravenous anifrolumab basic regimen (BR, 300 mg), intensified regimen (IR, 900 mg ×3, 300 mg thereafter) or placebo, alongside standard therapy (oral glucocorticoids, mycophenolate mofetil). The primary endpoint was change in baseline 24-hour urine protein–creatinine ratio (UPCR) at week (W) 52 for combined anifrolumab versus placebo groups. The secondary endpoint was complete renal response (CRR) at W52. Exploratory endpoints included more stringent CRR definitions and sustained glucocorticoid reductions (≤7.5 mg/day, W24–52). Safety was analysed descriptively.
Results Patients received anifrolumab BR (n=45), IR (n=51), or placebo (n=49). At W52, 24-hour UPCR improved by 69% and 70% for combined anifrolumab and placebo groups, respectively (geometric mean ratio=1.03; 95% CI 0.62 to 1.71; p=0.905). Serum concentrations were higher with anifrolumab IR versus anifrolumab BR, which provided suboptimal exposure. Numerically more patients treated with anifrolumab IR vs placebo attained CRR (45.5% vs 31.1%), CRR with UPCR ≤0.5 mg/mg (40.9% vs 26.7%), CRR with inactive urinary sediment (40.9% vs 13.3%) and sustained glucocorticoid reductions (55.6% vs 33.3%). Incidence of herpes zoster was higher with combined anifrolumab vs placebo (16.7% vs 8.2%). Incidence of serious adverse events was similar across groups.
Conclusion Although the primary endpoint was not met, anifrolumab IR was associated with numerical improvements over placebo across endpoints, including CRR, in patients with active lupus nephritis.
Trial registration number NCT02547922.
- autoimmune diseases
- biological therapy
- immune system diseases
- lupus erythematosus
- systemic
- lupus nephritis
Data availability statement
Data are available on reasonable request. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Deidentified participant data can be made available upon reasonable request to Catharina Lindholm (ORCHID ID: 0000-0002-0533-7185) or through the Vivli web-based data request platform. Reuse is permitted only with permission from AstraZeneca. The Clinical Study Protocol, Statistical Analysis Plan, and Clinical Study Report Synopsis are available at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=22674.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- autoimmune diseases
- biological therapy
- immune system diseases
- lupus erythematosus
- systemic
- lupus nephritis
Key messages
What is already known about this subject?
Anifrolumab is generally well tolerated and efficacious across a range of clinically meaningful endpoints in patients with systemic lupus erythematosus (SLE).
Anifrolumab targets the type I interferon signalling pathway, which plays a role in the pathogenesis of lupus nephritis (LN).
What does this study add?
This phase II, randomised, placebo-controlled trial is the first investigation of an interferon-targeted therapy in patients with active LN.
This study suggests that patients with LN require an intensified regimen (IR) of anifrolumab relative to non-renal SLE to obtain adequate exposure and clinical efficacy.
How might this impact on clinical practice or future developments?
The findings of TULIP-LN merit further investigation of anifrolumab IR in larger numbers of patients with active LN.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune condition that can cause multiorgan inflammation and organ damage.1 Lupus nephritis (LN) is one of the most prevalent severe disease manifestations of SLE, occurring in ~40% of patients.2 Patients with Class III or IV LN3 have poor prognoses, with up to 45% of patients progressing to end-stage kidney disease within 15 years of diagnosis.4–6
High type I interferon gene signatures (IFNGS) are present in >80% of patients with LN,7 become even more pronounced in active LN,8 and are associated with active kidney disease and treatment failure.8 9 Therefore, there is scientific rationale to support anifrolumab, a human monoclonal antibody that binds to the type I interferon receptor subunit 1,10 as a potential LN treatment option.
Anifrolumab has been investigated in patients with moderate to severe SLE despite standard therapy in two phase III randomised placebo-controlled trials, TULIP-1 and TULIP-2.11 12 Anifrolumab 300 mg was generally well tolerated and provided therapeutic benefit across several clinical endpoints despite TULIP-1 not meeting its primary endpoint.11 12 As the TULIP trials excluded patients with severe, active LN, further studies were required to evaluate anifrolumab in this patient population.11 12 Here, we report 52-week primary analysis results of the 2-year, phase II, randomised, placebo-controlled Treatment of Uncontrolled Lupus via the Interferon Pathway - Lupus Nephritis (TULIP-LN) trial, which evaluated the safety and efficacy of two anifrolumab dosages added to standard therapy in patients with active LN.
Methods
Study design
This phase II trial was conducted at 66 sites in 16 countries (online supplemental table S1) in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice Guideline. All patients provided written informed consent. The trial consisted of a 52-week randomised, placebo-controlled, double-blind treatment period, after which the primary endpoint was assessed. Patients then either entered an 8-week safety follow-up period or, if eligible, the ongoing second-year treatment period (online supplemental figure S1). Only the first-year data are reported here.
Supplemental material
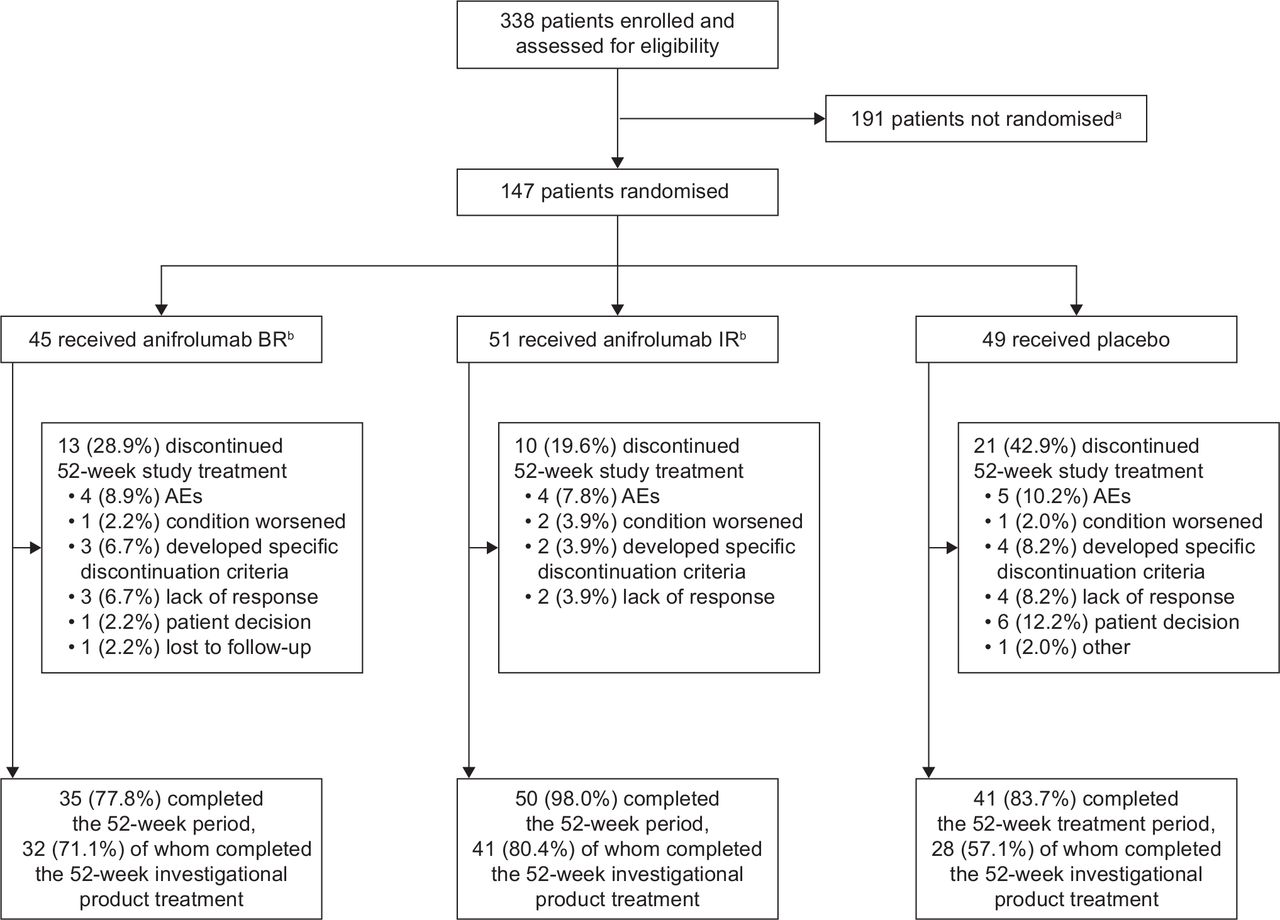
Patient disposition for the completed 52-week double-blind treatment period. All percentages are based on the 145 patients in the full analysis set (modified intention-to-treat population), who were included in the primary endpoint analysis. aOf patients not randomised, 179 did not meet the screening criteria, 7 withdrew consent, 2 experienced AEs, 1 was lost to follow-up, 1 patient was not included because of the physician’s decision, and 1 patient was not included for unspecified reason (‘other’). bOne patient was assigned to but did not receive ≥1 dose of each of the anifrolumab regimens and therefore was not included in the analysis. AE, adverse event; BR, basic regimen; IR, intensified regimen.
Patients
Eligible patients were 18–70 years old with a biopsy-proven diagnosis within 3 months of screening of Class III or IV (+/−coexistent Class V) LN, according to the WHO or International Society of Nephrology and the Renal Pathology Society (ISN/RPS) 2003 criteria.3 Eligible patients had 24-hour urine−protein creatinine ratios (UPCR) >1 mg/mg (113.17 mg/mmol), estimated glomerular filtration rates (eGFR) ≥35 mL/min/1.73 m2, and fulfilled ≥4 of the 11 American College of Rheumatology SLE 1997 classification criteria, including seropositivity for ≥1 of antinuclear, anti-double-stranded DNA (anti-dsDNA), and/or anti-Smith antibodies at screening.13 For full inclusion/exclusion criteria, see online supplemental material.
Treatments
Patients were block randomised (1:1:1) to receive anifrolumab basic regimen (BR; 300 mg, corresponding to SLE dosing10–12), anifrolumab intensified regimen (IR; 900 mg for the first three doses, 300 mg thereafter), or placebo intravenously every 4 weeks for 48 weeks. Randomisation was stratified according to 24-hour UPCR at screening (≤3.0 vs >3.0 mg/mg) and type I IFNGS status (high vs low, determined as previously described14).
Investigational agents were administered alongside standard therapy of oral glucocorticoids and mycophenolate mofetil (MMF). All patients received an intravenous methylprednisolone pulse (500 mg) within 10 days of randomisation. There was a mandatory oral glucocorticoid taper to a dosage goal of ≤10 mg/day by week 12 and ≤7.5 mg/day by week 24 (prednisone or equivalent). MMF was titrated to a target dosage of 2 g/day by week 8. MMF dosage adjustments were permitted for suboptimal responses, toxicity or intolerability. Stable oral glucocorticoid and MMF dosages were required during weeks 40–52. Standard therapy requirements are detailed further in online supplemental material.
Prespecified discontinuation criteria
During the 52-week treatment period, patients were required to discontinue investigational product treatment if they had predefined worsening of LN, which was defined as an LN-related, confirmed eGFR decrease >30% from baseline to <60 mL/min/1.73 m2 at any time, eGFR decrease <75% from baseline to <60 mL/min/1.73 m2 at week 12 or week 24, or nephrotic range UPCR at week 12 or week 24 (>3.5 mg/mg or <60% improvement in patients >3 mg/mg at baseline).
Investigational product was discontinued in the case of failure to adhere to protocol-specified standard therapy requirements, including a mandatory oral glucocorticoid taper to a dosage of ≤15 mg/day by week 12 or <15 mg/day by week 24. Patients were also required to discontinue investigational product treatment if they received rescue treatments (eg, cyclophosphamide, high-dose glucocorticoids and/or rituximab) owing to worsening LN or SLE at any time, or if they received protocol-specified forbidden medications at any time. Standard therapy requirements and forbidden medication rules are detailed further in online supplemental material.
Outcomes
Primary endpoint
The primary endpoint was the relative difference in the mean change from baseline to week 52 in 24-hour UPCR in the combined anifrolumab (IR plus BR) versus placebo group, measured with a geometric mean (GM) ratio (GMR; <1 favours anifrolumab) using the equation:

Secondary endpoint
The secondary endpoint was the difference in the combined anifrolumab vs placebo groups in the proportion of patients with a complete renal response (CRR) at week 52, defined as 24-hour UPCR ≤0.7 mg/mg, eGFR ≥60 mL/min/1.73 m2 or no decrease ≥20% from baseline, no investigational product discontinuation and no use of restricted medications. Restricted medications are listed in online supplemental material.
Exploratory endpoints
Exploratory endpoints included mean UPCR over time; the proportion of patients with sustained oral glucocorticoid tapers (≤7.5 mg/day prednisone equivalent from weeks 24–52, among those receiving ≥20 mg/day at baseline); the proportion of patients with an alternative CRR (aCRR), defined as a CRR that required inactive urine sediment (<10 red blood cells per high-power field); the proportion of patients with a CRR and sustained oral glucocorticoid taper; mean change from baseline in non-renal SLE Disease Activity Index 2000 (SLEDAI-2K),15 Physician’s Global Assessment (PGA),16 Patient’s Global Assessment (PtGA),17 lupus serologies (anti-dsDNA antibodies, C3/C4); and the immunogenicity, pharmacokinetic (PK) and pharmacodynamic (PD) profile of anifrolumab. PD neutralisation was measured as the median percentage change of baseline 21-gene type I IFNGS (21-IFNGS), as described previously.10 14 18
Post hoc analyses included cumulative proteinuria (area under the curve in UPCR standardised by expected follow-up time), the proportion of patients with a CRR with UPCR ≤0.5 mg/mg (CRR0.5), and probability of CRR0.5 response sustained through week 52.
Safety assessments included adverse events (AEs), laboratory assessments and vital signs. AEs of special interest (AESI) were non-opportunistic serious infections, opportunistic infections, herpes zoster (HZ), influenza, malignancy, tuberculosis, hypersensitivity and major adverse cardiovascular events.
Sample size estimation
A 1:1:1 randomised sample size of 50 patients per treatment arm was planned to provide ~87% power at the two-sided alpha level of 0.0499 to detect a relative difference of 0.76 or less in 24-hour UPCR GMR from baseline to week 52 for combined anifrolumab versus placebo.
Statistical analysis
The primary endpoint was analysed using a mixed model for repeated measures fitted to log-transformed 24-hour UPCR values, controlling for stratification factors and based on observed data up to investigational product discontinuation. Binary endpoints, responder rates and 95% CIs were calculated using a stratified Cochran-Mantel-Haenszel approach, controlling for stratification factors. Safety was analysed descriptively.
Efficacy and safety analyses were conducted using the modified intention-to-treat (mITT) population. Patients enrolled at sites in Italy and France were excluded from the analyses of secondary and exploratory binary CRR efficacy endpoints. This exclusion was because the Italian Medicines Agency and the France Ethics Committee did not agree to a protocol amendment that included changes to the cut-off values for the renal function and proteinuria components of the CRR definition.
All analyses were performed with Statistical Analysis System (SAS; SAS Institute Inc., Cary, NC), V.9.3 or higher. Individual anifrolumab regimens versus placebo analyses were conducted using a hierarchical testing strategy to control the familywise error. Further details on statistical analyses are provided in online supplemental material.
Patient and public involvement
Patients and/or the public were not involved in the design, conduct, reporting or dissemination of this research.
Results
Trial population
Between November 2015 and November 2018, 338 patients were screened, and 147 patients were randomised (figure 1). Of the 145 patients in the mITT population, 45 received anifrolumab BR, 51 received anifrolumab IR and 49 received placebo.
Demographics and baseline disease characteristics are shown in table 1. At screening, 26.9% of patients had Class III LN and 73.1% of patients had Class IV LN (41.0% and 21.7% of whom had coexistent Class V disease, respectively). Most patients (94.5%) were IFNGS high. At baseline, 77.2% of patients had eGFR ≥60 mL/min/1.73 m2. Demographics and baseline disease characteristics were generally balanced between groups; however, the placebo group had higher mean baseline 24-hour UPCR, lower mean baseline eGFR, longer median time from initial LN diagnosis and more patients with low C3 or C4 than both anifrolumab groups. Most patients were receiving standard therapy for LN at baseline (mean dosage 22.3 mg/day prednisone equivalent oral glucocorticoids and 1.8 g/day MMF); treatments were balanced between groups.
Patient demographics and disease characteristics
Overall, 126/145 patients (86.9%) completed the 52-week period (BR: 77.8%, IR: 98.0%, placebo: 83.7%), and 101/145 patients (69.7%) completed investigational product treatment at week 52 (figure 1). More patients discontinued investigational product early in the placebo (42.9%) than in both anifrolumab groups (BR: 28.9%, IR: 19.6%; online supplemental figure S2). There were 75 patients who entered the second-year extension period; only the first-year results are reported here.
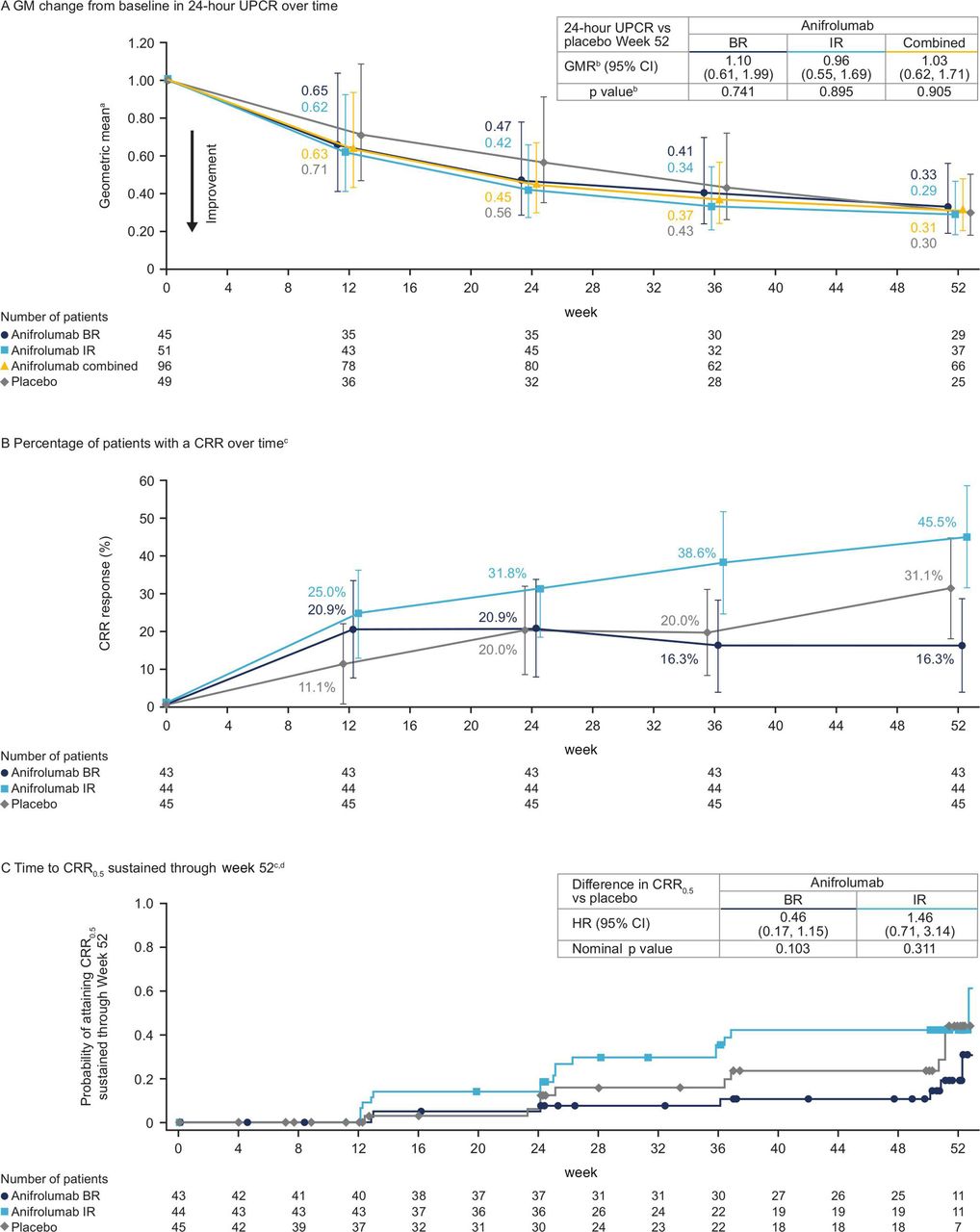
Key efficacy endpoints over time. Error bars represent 95% CIs. aGM of the ratio of the 24-hour UPCR values at each time point over the baseline value for each treatment group (values <1 indicate an improvement). bGMR of the relative improvement in 24-hour UPCR for anifrolumab groups vs placebo groups, where GMR <1 favours anifrolumab. A p≤0.05 for the combined anifrolumab vs placebo group was deemed significant. All other p values presented are nominal. cPatients from France and Italy (n=13) were excluded from the analysis (see online supplemental material). dProbability of obtaining a sustained CRR0.5 was analysed post hoc using a Cox regression model controlling for stratification factors. BR, basic regimen; CRR, complete renal response; CRR0.5, CRR with UPCR ≤0.5 mg/mg; GM, geometric mean; GMR, geometric mean ratio; IR, intensified regimen; UPCR, urine protein–creatinine ratio.
Efficacy
Primary endpoint
The primary endpoint was not met; at week 52, the mean 24-hour UPCR improved from baseline by 69% and 70% to 0.92 mg/mg and 1.05 mg/mg in the combined anifrolumab and placebo groups, respectively, resulting in a GMR of 1.03 (95% CI 0.62 to 1.71, p=0.905; GMR <1 favours anifrolumab; figure 2A; online supplemental table S2). The outcome of the preplanned efficacy analysis for the combined anifrolumab versus placebo group was made less clinically meaningful by the suboptimal PK exposure and PD neutralisation in the anifrolumab BR group, owing to higher drug clearance associated with proteinuria in patients with active LN versus patients with non-renal SLE19 20 (see later sections). As the primary endpoint was not met, the secondary endpoint was not formally tested per the statistical analysis plan. For all remaining endpoints, reported p values are nominal and should not be used to conclude statistical significance.
Mean 24-hour UPCR improved over time across treatment groups (online supplemental figure S3). The GM improvements in 24-hour UPCR were numerically larger in both anifrolumab groups vs placebo at weeks 12 and 24, but not at weeks 36 or 52 (figure 2A). In the anifrolumab IR group, the 24-hour UPCR improved by 71% from baseline to 0.88 mg/mg at week 52, which was similar to the improvement with placebo (GMR=0.96; 95% CI 0.55 to 1.69) (figure 2A). Both anifrolumab groups had numerically lower cumulative UPCRs than placebo throughout the treatment duration (online supplemental figure S4). There were no major differences in 24-hour UPCR changes from baseline to week 52 across predefined subgroups (online supplemental figure S5). Post hoc sensitivity analysis controlling for time from LN diagnosis did not reveal any major impact on primary results (data not shown).

{kind=link}
{kind=link}
{kind=link}
Measures of disease activity and IFNGS neutralisation over time. Number of patients with non-missing value at visit are presented. SLEDAI-2K, PGA and PtGA change from baseline were analysed using a mixed model for repeated measures, controlling for stratification factors, and based on observed data up to investigational product discontinuation. PD neutralisation was analysed descriptively. BR, basic regimen; IFN, interferon; IFNGS, interferon gene signature; IR, intensified regimen; LS, least squares; MAD, median absolute deviation; PD, pharmacodynamic; PGA, Physician’s Global Assessment, PtGA, Patient’s Global Assessment; SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity Index 2000.
Secondary endpoint
At week 52, the percentages of patients with a CRR were similar in the combined anifrolumab and placebo groups (31.0% vs 31.1%, difference −0.1% (95% CI −16.9 to 16.8)) (table 2). The proportion of patients with a CRR was greater in the anifrolumab IR group than in the placebo group (45.5% vs 31.1%, difference 14.3% (95% CI −5.8 to 34.5)) and was lower in the anifrolumab BR group than in the placebo group (16.3% vs 31.1%, difference −14.8% (95% CI −32.9 to 3.2)).
Summary of secondary and exploratory endpoints
The proportions of patients in each treatment group who attained the individual components of the CRR at week 52 are displayed in online supplemental table S3. A greater proportion of patients in the anifrolumab IR group had 24-hour UPCR ≤0.7 mg/mg at week 52 compared with the anifrolumab BR or placebo groups (anifrolumab IR: 50.0%; anifrolumab BR: 32.6%; placebo: 35.6%). At week 52, 81.8% of the anifrolumab IR group, 79.1% of the anifrolumab BR group, and 73.3% of the placebo group had eGFR ≥60 mL/min/1.73 m2 or no decrease ≥20% from baseline, with mean (SD) eGFR values of 94.5 (36.2), 95.8 (24.9) and 84.7 (30.1) mL/min/1.73 m2, respectively.
Exploratory endpoints
The proportion of patients who had an aCRR at week 52 (which required inactive urinary sediment) was greater in the anifrolumab IR group than the placebo group (40.9% vs 13.3%, difference 27.6% (95% CI 9.4 to 45.7)), and was lower in the anifrolumab BR group than in the placebo group (7.0% vs 13.3%, difference −6.4% (95% CI –20.6 to 7.8)). The proportion of patients with inactive urinary sediment (<10 red blood cells per high-power field) was also greater in the anifrolumab IR group (77.3%) than in the anifrolumab BR group (55.8%) or placebo group (42.2%) (online supplemental table S3).
A similar trend was observed with CRR0.5; the proportion of patients who had a CRR0.5 at week 52 was greater in the anifrolumab IR group than the placebo group (40.9% vs 26.7%, difference 14.2% (95% CI −5.4 to 33.9)) and was lower in the anifrolumab BR group than in the placebo group (16.3% vs 26.7%, difference −10.4 (95% CI −28.1 to 7.3) (table 2).
Response rates were higher with anifrolumab IR vs placebo as early as week 12 and remained higher over time across all CRR definitions (figure 2B; online supplemental figure S6). Compared with placebo, patients in the anifrolumab IR group were more likely to have a CRR0.5 response sustained through week 52 (sustained CRR0.5 HR 1.46; 95% CI 0.71 to 3.14) (figure 2C). Anifrolumab BR responses for all CRR definitions were generally similar to or lower than placebo at all timepoints apart from week 12 (figure 2B, online supplemental figure S6).
The proportion of patients who had a sustained oral glucocorticoid dosage taper ≤7.5 mg/day was greater in the anifrolumab IR group than in the placebo group (55.6% vs 33.3%, difference 22.2% (95% CI −0.8 to 45.2)) and was similar in the anifrolumab BR and placebo groups (35.5% vs 33.3%, difference 2.2% (95% CI −21.4 to 25.7)). The proportion of patients who had a CRR with sustained glucocorticoid taper was also greater in the anifrolumab IR group than in the placebo group (34.1% vs 24.4%, difference 9.7% (95% CI −9.5 to 28.8)) but was lower in the anifrolumab BR group than in the placebo group (14.0% vs 24.4%, difference −10.5 (95% CI –27.6 to 6.6)) (table 2).
Compared with placebo, anifrolumab IR was associated with greater improvements from baseline in measures of disease activity (SLEDAI-2K, PGA and PtGA), whereas the anifrolumab BR was associated with greater improvements in SLEDAI-2K but not for PGA or PtGA (figure 3A−C). Improvements from baseline in lupus serologies (anti-dsDNA antibodies, C3 and C4) were variable; however, there was a trend towards greater improvements in anti-dsDNA antibodies and C3 levels with anifrolumab IR and anifrolumab BR than with placebo by week 52 (online supplemental figure S7).
Pharmacokinetics
The PK analysis included 95 patients who received anifrolumab and had ≥1 quantifiable serum PK observation after the first dose. Anifrolumab exhibited non-linear PK between the anifrolumab BR and IR groups (online supplemental figure S8). In IFNGS-high patients (94.5%), the median week 12 anifrolumab steady-state concentration was 63.4 µg/mL with anifrolumab IR and 8.2 µg/mL with anifrolumab BR (~50% lower than in non-renal SLE21) (online supplemental figure S9). After anifrolumab IR was tapered to 300 mg at week 12, the median trough concentrations at week 24 and week 36 were lower than in patients with non-renal SLE. Anifrolumab clearance was higher among patients with UPCR >3 mg/mg vs ≤3 mg/mg at baseline (online supplemental figure S10). Anifrolumab clearance decreased over time. Larger decreases in baseline clearance (≥33% decrease at week 52) were associated with greater reductions in baseline 24-hour UPCR after week 12 compared with patients who had smaller decreases in baseline clearance (<20% decrease at week 52) (online supplemental figure S11). This association was observed to a greater extent in patients with baseline 24-hour UPCR >3 mg/mg (who had higher clearance) compared with patients with baseline 24-hour UPCR ≤3 mg/mg (online supplemental figure S11).
Pharmacodynamics
The PD analysis included 137 IFNGS-high patients. A median PD neutralisation >80% was observed with anifrolumab IR across all visits (weeks 12, 24, 36 and 52). Sustained PD neutralisation to this degree was not observed with anifrolumab BR (figure 3D). Minimal PD neutralisation was observed in the placebo group.
Safety and tolerability
Table 3 shows the safety summary. The percentages of patients with any AE were 95.6%, 92.2% and 89.8% in the anifrolumab BR, anifrolumab IR and placebo groups, respectively. The AEs that were more common (≥5% difference) in the combined anifrolumab versus placebo groups were HZ, urinary tract infection and influenza. Serious AEs occurred in 22.2%, 17.6% and 16.3% of the anifrolumab BR, anifrolumab IR and placebo groups, respectively. HZ was the only serious AE reported in >1 patient per treatment group. There were no deaths during the treatment period. There was one fatal vascular neurological AE in the anifrolumab BR group during the follow-up. AEs leading to investigational product discontinuation occurred in 11.1%–12.2% of patients across groups.
AEs during the treatment period (mITT population)
Overall, AESIs occurred in 24.0% and 16.3% of patients in the combined anifrolumab and placebo groups, respectively. Of the AESIs, HZ and influenza occurred more commonly in the combined anifrolumab versus placebo group. HZ occurred in 20.0%, 13.7% and 8.2% of patients in the anifrolumab BR, anifrolumab IR and placebo groups, respectively. Of the 16 HZ cases in the combined anifrolumab group, the majority were of mild to moderate intensity, 6 were serious, and all were cutaneous (13 localised, 3 disseminated). HZ events tended to occur early in the trial (online supplemental figure S12) and were resolved with conventional treatment. The incidence of other AESIs was low across groups.
Discussion
There is high unmet need in the treatment of LN. Despite recent advances, remission rates remain suboptimal,22–24 and patients are at high risk of developing end-stage kidney disease4–6 and drug-related toxicity, particularly relating to prolonged, high-dose glucocorticoid use.19 25
Here, we report the primary analysis results of the phase II TULIP-LN trial, which explored the safety and efficacy of two anifrolumab dosing regimens alongside standard therapy in patients with active LN. The primary endpoint was not met; however, UPCR improvement in the combined anifrolumab group versus placebo group was adversely impacted by the suboptimal anifrolumab exposure obtained with BR dosing (~50% lower than in non-renal SLE21). The suboptimal PK exposure with anifrolumab BR was likely related to increased clearance associated with proteinuria in LN19 20; indeed, we observed an association between the magnitude of decrease in anifrolumab clearance and the improvement in 24-hour UPCR over time. The suboptimal PK exposure obtained with the anifrolumab BR regimen was also reflected in the lower degree of 21-IFNGS neutralisation and relatively infrequent clinical responses observed with anifrolumab BR. The anifrolumab IR was required to attain serum exposure and PD neutralisation that was similar to levels observed in non-renal SLE.26 As such, the anifrolumab IR was required to reach clinical efficacy, with clinically meaningful responses across renal endpoints, including proteinuria, multiple stringent CRR definitions (including requirements for UPCR ≤0.5 mg/mg or inactive urinary sediment), sustained oral glucocorticoid dosage reductions, disease activity measures and lupus serologies.
Reduction of proteinuria is associated with reduced risk of end-stage kidney disease27–29; thus, it is an appropriate and objective surrogate endpoint for a proof-of-concept trial. Here, numerically greater improvements in 24-hour UPCR were observed early in the trial with both anifrolumab groups vs placebo; however, by week 52, all treatment groups had improvements in baseline 24-hour UPCR of approximately 70%. In the placebo group, the 24-hour UPCR improvement may have been overestimated, owing to large amounts of missing data generated from the high rate of investigational product discontinuation. These missing data were imputed into the primary analysis model; however, high levels of data imputation could confound the model-estimated treatment effect. In the cumulative UPCR analysis, treatment with both anifrolumab regimens numerically improved cumulative proteinuria over time more than placebo. By week 52, cumulative UPCR was ~30% and ~20% lower than placebo in the anifrolumab IR and BR groups, respectively. Cumulative UPCR signifies overall proteinuria improvement over time, so it may be less susceptible to short-term confounders, including collection errors, diet and exercise.30 31
Anifrolumab IR was also associated with clinically meaningful responses over placebo across CRR definitions as early as week 12, including the robust composite endpoint CRR0.5, which is favoured for registrational clinical trials.32 33 Anifrolumab IR yielded the strongest response (treatment difference 28%) for aCRR, a highly stringent endpoint requiring no haematuria (a pathognomonic marker of active glomerular inflammation34). More patients also achieved a sustained oral glucocorticoid dosage reduction and a CRR with a sustained dosage reduction with anifrolumab IR vs placebo, which merits further exploration, as reducing glucocorticoid dosages is a key treatment goal for patients with LN.19 25
The safety profile of anifrolumab in LN was generally consistent with SLE without active renal disease, including higher incidence of HZ with anifrolumab versus placebo.10 35 Most AEs were mild or moderate in intensity, were not serious, and did not lead to investigational product discontinuation.35 In alignment with previous observations,36–38 the incidence of HZ was higher among patients with LN than those with non-renal SLE. This was likely related to LN requiring more potent background immunosuppressive regimens, including glucocorticoids,19 which are identified risk factors for HZ reactivation.37 39 Consistently, HZ tended to occur early in the trial when glucocorticoids had not yet been tapered. Most HZ events were mild or moderate, cutaneous, and resolved with antivirals without investigational product discontinuation.
Limitations include that this was a proof-of-concept, dose-finding study, with a relatively small enrolment of patients. There was also a high rate of investigational product discontinuation; as discussed previously, this may have confounded the high 24-hour UPCR improvement estimate for placebo. Discontinuations could also have impacted binary response rates, as patients meeting the discontinuation criteria or using restricted medications were classified as non-responders, irrespective of disease activity improvements.
Overall, the TULIP-LN study results support further assessment of the efficacy and safety of anifrolumab IR in patients with active LN. The PK results suggest that three intensified doses of anifrolumab 900 mg improved clearance to a lower rate, enabling dosage tapering to the 300 mg every 4 weeks regimen indicated for patients with SLE without active renal disease.40 In patients with active LN, the anifrolumab IR was required to obtain clinical efficacy; indeed, the anifrolumab IR was numerically superior to placebo for several clinically relevant endpoints, whereas the anifrolumab BR was not. As such, the results suggest that the anifrolumab IR is a more suitable dosing regimen than the anifrolumab BR to carry forward into future clinical investigations of anifrolumab to treat patients with active LN. Learnings from this trial will support dose selection and the development of trial designs for future studies of anifrolumab in LN.
Data availability statement
Data are available on reasonable request. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Deidentified participant data can be made available upon reasonable request to Catharina Lindholm (ORCHID ID: 0000-0002-0533-7185) or through the Vivli web-based data request platform. Reuse is permitted only with permission from AstraZeneca. The Clinical Study Protocol, Statistical Analysis Plan, and Clinical Study Report Synopsis are available at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/View?id=22674.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol was approved by each centre’s ethics committee or institutional review board. The study was overseen by an external data and safety monitoring board. Please refer to uploaded file named '221659 RP06_IRB_EC Submission and Approval_20210818' for all Approval Numbers/IDs. Of note, France and Italy rejected Protocol Amendment number 3; therefore, patients from France and Italy are excluded from all relevant analyses. This is clearly specified in the manuscript where applicable. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank the investigators, research staff, healthcare providers and especially the patients who participated in this study. We would also like to acknowledge Micki Hultquist and the anifrolumab clinical team, particularly William Gunther and Jacek Gregorczyk. We would also like to acknowledge Frederick Jones for programmatic support, Gabriel Abreu for statistical support, and Gabor Illei for scientific support. Medical writing support was provided by Matilda Shackley, MPhil, of JK Associates, part of Fishawack Health.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors DJ, RAF, FAH, TT, RT and CL conceived and designed the study. BR, RAF, TT, JK, ES and RT acquired the data. All authors analysed and interpreted the data. All authors were involved in development, review and final approval of the manuscript. CL is the author acting as guarantor
Funding This support was funded by AstraZeneca.
Competing interests DJ received grants or contracts from GlaxoSmithKline, consultancy fees from AstraZeneca, Boehringer-Ingelheim, Chemocentryx, GlaxoSmithKline, Novartis, Roche, Takeda and Vifor, speaker fees from Amgen, GlaxoSmithKline and Vifor, and owns stocks in Aurinia. BR received consulting fees from Aurinia, AstraZeneca, Calliditas, Tavere, Novartis, Omeros, Chemocentryx, Morphosys, Bristol Myers Squibb, and Janssen. EM received consulting fees from Pfizer, AbbVie, GlaxoSmithKline, Sandoz, Eli Lilly, Bristol Myers Squibb and AstraZeneca, speaking fees from Pfizer, Amgen, AbbVie, Eli Lilly and Roche, and honoraria from Pfizer, AbbVie and Eli Lilly. RAF received consulting fees, payment or honoraria, and support for attending meetings and/or travel from AstraZeneca, and has participated on a Data Safety Monitoring Board or Advisory Board for AstraZeneca. FAH has received grants and consulting fees from and has participated on a Data Safety Monitoring Board or Advisory Board for GlaxoSmithKline, and has received consulting fees from Idorsia. TT, JK, ES, RT and CL are employees of and own shares of AstraZeneca. YLC is a former employee of and owns shares of AstraZeneca and a current employee of Seagen.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.