Article Text

Abstract
Objective: To investigate the significance of complement activation in patients with primary antiphospholipid syndrome (APS).
Methods: Thirty-six patients with primary APS, 42 control patients with non-systemic lupus erythematosus (SLE) connective tissue diseases, and 36 healthy volunteers were analysed retrospectively. Serum complement levels (C3, C4, CH50) and anaphylatoxins (C3a, C4a, C5a) were examined in all subjects, and serum complement regulatory factors (factor H and factor I) were measured in patients with primary APS. Plasma anticoagulant activity was determined in a mixing test using the activated partial thromboplastin time.
Results: Serum complement levels were significantly lower in patients with primary APS than in patients with non-SLE connective tissue diseases (mean (SD) C3: 81.07 (17.86) vs 109.80 (22.76) mg/dl, p<0.001; C4: 13.04 (8.49) vs 21.70 (6.96) mg/dl, p<0.001; CH50: 31.32 (8.76) vs 41.40 (7.70) U/ml, p<0.001) or healthy volunteers. Only two healthy subjects with low serum C4 levels showed hypocomplementaemia, whereas most patients with primary APS showed raised serum C3a and C4a. No subjects showed raised C5a. Patients with primary APS with low serum C3 or C4 had significantly higher levels of C3a or C4a than healthy controls. No patients had low serum complement regulatory factors. Among patients with primary APS, hypocomplementaemia was significantly more common in those with high anticoagulant activity than in those with low or normal activity.
Conclusion: Hypocomplementaemia is common in patients with primary APS, reflecting complement activation and consumption, and was correlated with anticoagulant activity, suggesting that antiphospholipid antibodies may activate monocytes and macrophages via anaphylatoxins produced in complement activation.
Statistics from Altmetric.com
Antiphospholipid syndrome (APS) is a clinical condition characterised by recurrent venous/arterial thrombosis and pregnancy morbidity in the presence of antiphospholipid antibodies (aPL).1 Despite the strong association between aPL and clinical manifestations, the pathogenic role of aPL has not been fully elucidated2; however, this role is possibly multifactorial in nature. The engagement of aPL on cell surfaces promotes intracellular signalling3 4; in endothelial cells, this leads to expression of procoagulant activity and adhesion molecules5 that in turn increases leukocyte adherence to endothelial cells.6 In monocytes, it leads to the upregulation of tissue factor7 8 that can enhance thrombin-induced platelet activation/aggregation.9 Thrombosis is also favoured by aPL-induced depression of fibrinolysis10 and by aPL interference with natural anticoagulant activities.11–13
Complement is part of the innate immune system and represents one of the effector arms of antibody-mediated immunity.14 The complement system is commonly activated in systemic lupus erythematosus (SLE) and is strongly associated with the physiopathology of inflammation, as suggested by the low serum complement concentration with increased deposition at sites of tissue damage. Complement-derived inflammatory mediators (anaphylatoxins) such as C3a, C4a and C5a increase vascular permeability, activate platelets15 and neutrophils,16 and promote release of cytokines such as tumour necrosis factor (TNF) α from monocytes,17 with simultaneous induction of systemic inflammation and coagulation.
A number of studies on murine models have highlighted how complement activation is essential for aPL-induced pregnancy morbidity.18 19 C5a, the most powerful inflammatory anaphylatoxin, seems to be crucial in clinical manifestation in these models.18 These findings have provided a new insight, suggesting that tissue injury in APS may be caused by a complement-mediated inflammatory process, rather than by thrombosis alone.20 Since complement activation in aPL-related thrombosis has not been examined clearly, this study was performed to evaluate the prevalence and relevance of hypocomplementaemia in patients with primary APS.
PATIENTS AND METHODS
Patients
This study was performed with a retrospective and cross-sectional design and included 36 patients with primary APS treated at Hokkaido University Hospital from 1996 to 2006. Primary APS was diagnosed according to the classification criteria for APS,1 21 with exclusion of patients who fulfilled criteria for SLE.22 Thirty-six age and gender-matched healthy volunteers and 42 non-SLE patients with connective tissue disease (15 systemic sclerosis, 9 Sjögren’s syndrome, 11 polymyositis/dermatomyositis, 5 mixed connective tissue disease, 1 overlap syndrome and 1 allergic granulomatous angiitis) were enrolled as controls. The non-SLE connective tissue disease control group comprised consecutive patients attending our autoimmune disease and rheumatology clinic who matched as controls and agreed to join this study. None of the participants had complications associated with infection, malignancy, impaired circulation or tissue ischaemia at the time of blood collection and all were negative for C-reactive protein.
Clinical records were reviewed retrospectively or patients were interviewed at the time of blood sample collection, or both. Arterial events such as stroke, myocardial infarction and iliac artery occlusion were confirmed by CT scan, magnetic resonance imaging (MRI) or angiography, as required. Deep vein thrombosis and pulmonary thrombosis were defined as venous thrombosis and confirmed by angiography or scintigraphy. Clinical manifestations of APS were diagnosed by rheumatologists with reference to imaging tests and clotting assays.
Plasma and serum sample collection
Blood was drawn by venepuncture and collected into different tubes. Blood samples for clotting assays were collected in plastic tubes containing 0.105 M citrate, immediately centrifuged at 3000 rpm for 15 min at 4°C, filtered through a 0.22 μM pore size membrane to obtain platelet-free plasma, and stored at −80°C until use. In the patients with primary APS, the plasma levels of d-dimer and fibrin degradation product (FDP) were determined using LPIA ace d-dimer II (Mitsubishi Kagaku Iatron, Tokyo, Japan) and Nanopia P-FDP (Daiichi Pure Chemical Co, Tokyo, Japan) kits, respectively. Blood samples for anaphylatoxin determination were collected in EDTA tubes containing nafamostate mesilate and centrifuged immediately to avoid cold in vitro complement activation.23 Blood samples for measurement of serum complement were drawn in plain tubes, allowed to clot and then centrifuged before measurement.
The study was done in accordance with the Declaration of Helsinki and the Principles of Good Clinical Practice. Approval was obtained from the local ethics committee, and informed consent was obtained from each subject before enrolment.
aPL determination
The levels of IgG/M anticardiolipin (aCL) and IgG/M anti-β2-glycoprotein I (β2GPI) antibodies were measured using a standard aCL ELISA24 and an in-house ELISA assay,25 respectively. The positive cut-off values of the assays were set at the 99th centile for 134 healthy controls, according to laboratory criteria for APS.1 IgG/M phosphatidylserine-dependent antiprothrombin (aPS/PT) antibodies were assayed as previously described.26 Lupus anticoagulant (LA) was determined by two clotting assays using an opto-mechanical coagulation analyser (Start4, Diagnostica Stago, Asnières, France) based on the guidelines of the Subcommittee on Lupus Anticoagulant/Phospholipid-Dependent Antibody.27 For the activated partial thromboplastin time (aPTT), a sensitive reagent with a low phospholipid concentration (PTT-LA, Diagnostica Stago) was used for screening, with confirmation using a Staclot LA kit (Diagnostica Stago). The dilute Russell’s viper venom time was determined using a Gradipore LA test (Gradipore, Frenchs Forest, NSW, Australia). LA was considered positive when at least one of these tests was positive for LA, and was arbitrarily classified into either strong or weak LA according to the anticoagulant potential, as follows. One volume of sample plasma was mixed with four volumes of normal pooled platelet-free plasma, and the clotting time of the mixture was measured using PTT-LA. LA was defined as strong if the aPTT ratio (1:4 mixed plasma/normal plasma) was >1.28 and weak if this ratio was <1.28.
Serum complement and anaphylatoxin determination
Complement components C3, C4 and C5 were determined by a nephelometric method that gives normal ranges of 86–160, 17–45 and 9–13 mg/dl, respectively. CH50 activity was determined by the Mayer method, with a normal range of 30–45 U/ml. Serum anaphylatoxin levels were determined by radioimmunoassay (complement C3a des-Arg [125I], complement C4a des-Arg [125I], complement C5a des-Arg [125I], Human Assay, GE Healthcare Bioscience, London, UK), with normal ranges of 50–200 ng/ml for C3a, 50–250 ng/ml for C4a and <10 ng/ml for C5a. C3, C4 and CH50 were measured in all participants, C5 was determined in 10 patients with primary APS, and anaphylatoxins were examined in 17 patients with primary APS, 9 control non-SLE patients and 17 healthy controls.
Plasma TNFα level determination
Plasma TNFα levels were examined in 22 patients with APS using sandwich ELISA (Endogen Human TNFα ELISA kit: Pierce Biotechnology, Rockford, Illinois, USA).
Measurement of serum complement regulatory factor
Serum levels of complement regulatory factor H (C3b-related C5 activation inhibitor) and factor I (C4b-related C3 activation inhibitor) were measured in 16 and 13 patients with APS, respectively, by radioimmunoassay (Monoclonal Antibody to Human Factor H, Monoclonal Antibody to Human Factor I, Quidel Corporation, San Diego, California, USA). The normal ranges are 22.8–41.7 mg/dl for factor H and 3.3–14.4 mg/dl for factor I, according to the manufacturer’s instructions.
Measurement of serum immune complex
Serum levels of immune complex were measured in 33 patients with primary APS and 22 patients with non-SLE connective tissue disease, by enzyme immunoassay (Immunocomplex mRF Nissui, Nissui Pharmaceutical, Tokyo, Japan). The normal range is <4.2 μg/ml, according to the manufacturer’s instruction.
Statistical analysis
A Student t test, Mann–Whitney non-parametric test, Pearson correlation coefficient or Fisher’s exact test was used as appropriate. In categorical analysis, the relative risks were expressed as odds ratios with 95% confidence intervals (95% CI). SPSS II for Windows was used for all calculations.
RESULTS
Serum complement levels and prevalence of hypocomplementaemia in primary APS
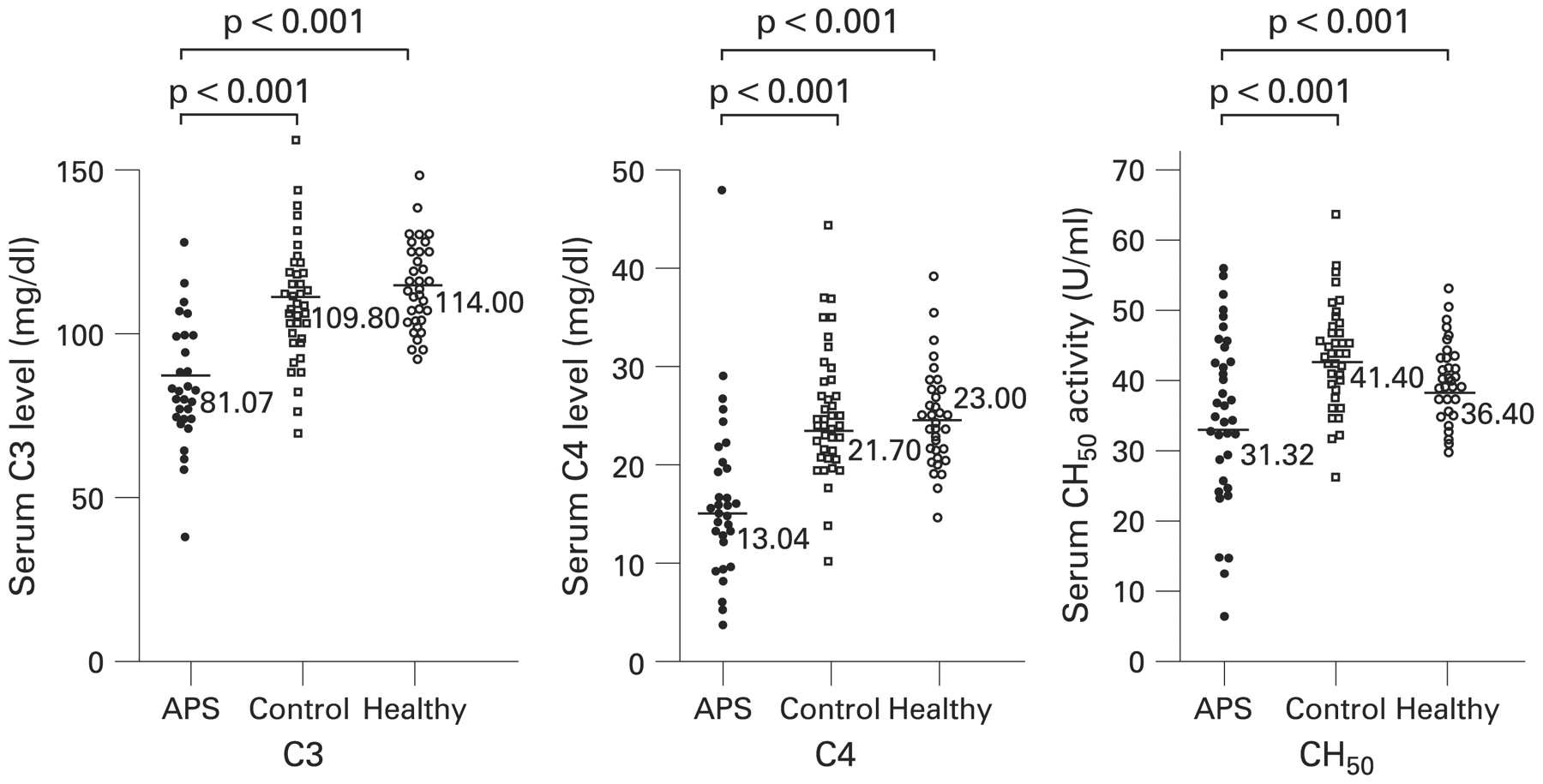
Criteria for SLE were investigated in the subjects by chart review or interview of the patients. The number of lupus criteria met by the primary APS group did not differ from that for patients with non-SLE connective tissue diseases (table 1). Signs of acute thrombosis were not detected in any patients with primary APS at the time of blood collection, and no significant increase of plasma d-dimer or FDP was found in these patients. The mean (SD) period after the last manifestation in patients with primary APS was 4.2 (3.6) years (range 0.3–10) (table 1). The patients with primary APS showed a higher prevalence of reduced levels of C3, C4 and CH50 than patients with non-SLE connective tissue diseases (C3: 69.4% vs 9.52%, OR = 21.59, 95% CI 6.18 to 75.42; C4: 61.1% vs 7.1%, OR = 15.32, 95% CI 4.48 to 52.31; CH50: 47.2% vs 2.4%, OR = 36.68, 95% CI 4.54 to 296.26). No healthy volunteers had a reduced complement level, but two had a low serum C4 level. The mean levels of C3, C4 and CH50 were lower in patients with primary APS than in patients with non-SLE connective tissue disease or healthy volunteers (fig 1). Additionally, serum complement levels of patients with non-SLE connective tissue disease with a past history of thrombosis (C3: 112 (13.5); C4: 20.0 (6.32); CH50: 40.4 (4.54)) did not show significant differences with the patients without a past history of thrombosis.

Serum complement levels. C3, C4 and CH50 levels in patients with primary antiphospholipid syndrome (APS), patients with non-systemic lupus erythematosus (SLE) connective tissue diseases and healthy volunteers. Control, patients with non-SLE connective tissue diseases; healthy, healthy volunteers. Statistical analysis by Student t test.
Serum anaphylatoxin levels
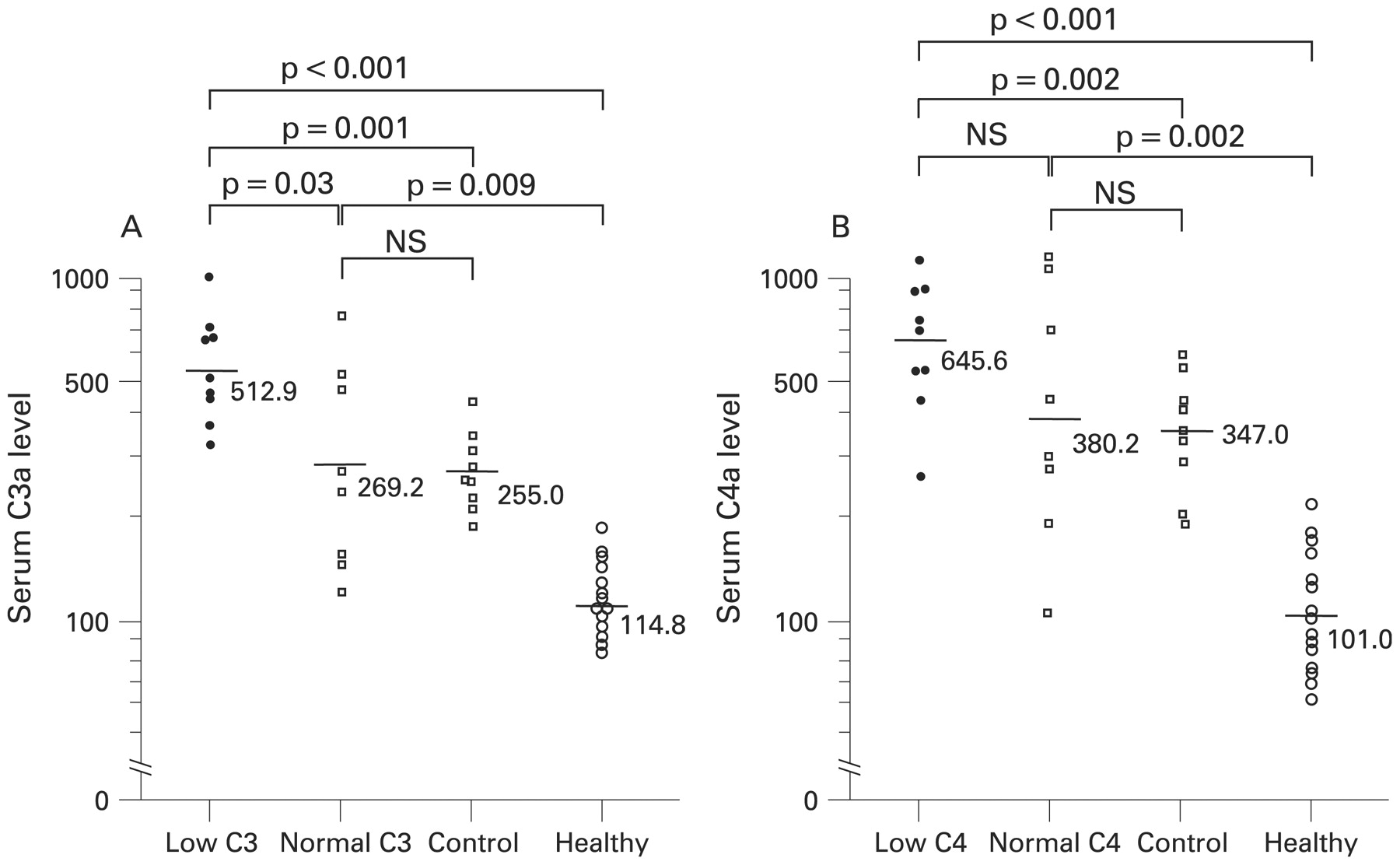
Most patients with primary APS showed raised serum C3a and C4a levels (C3a: 13/17, 76%; C4a: 14/17, 82%), but none showed raised C5a (0/17). Patients with primary APS with a low serum C3 concentration had significantly higher levels of C3a than those with normal C3, but there was no significant difference in C4a levels between patients with low and normal C4 (fig 2). The non-SLE patients had significantly lower levels of C3a and C4a than patients with primary APS with low serum C3 and C4 concentrations. However, there were no significant differences in C3a and C4a levels between control patients and patients with primary APS with normal C3 and C4 levels. C3a and C4a levels of control patients with past history of thrombosis (n = 4; C3a: 2 68 (73.1); C4a: 362 (62.1)) did not show significant differences from patients without a past history of thrombosis. No healthy volunteers showed raised serum anaphylatoxin levels.

Serum anaphylatoxin levels. Serum C3a and C4a levels were measured in 17 patients with primary antiphospholipid syndrome (APS), 9 patients with non-systemic lupus erythematosus (SLE) connective tissue diseases and 15 healthy volunteers. The bars and figures in the graphs represent the mean levels of anaphylatoxins in each groups. (A) Comparison of C3a levels between patients with primary APS with low or normal C3, control patients and healthy volunteers. (B) Comparison of C4a levels between patients with primary APS with low or normal C4, control patients and healthy volunteers. Control, patients with non-SLE connective tissue diseases; healthy, healthy volunteers. Statistical analysis by Student t test.
Plasma TNFα levels in patients with primary APS
Raised plasma TNFα was found in 7/22 (32%) of patients with primary APS. The prevalence of raised TNFα was greater in patients with hypocomplementaemia than in those with the normal serum CH50 activity (63% vs 14%, OR = 10, 95% CI 1.26 to 79.34).
Serum complement regulatory protein levels
Serum complement regulatory factor H and factor I were measured in 16 and 13 patients with primary APS, respectively. These patients did not have reduced levels of factor H or factor I (fig 3). Serum factor H levels tended to be raised in patients with high C3a serum levels, but the increase was not statistically significant (fig 3).
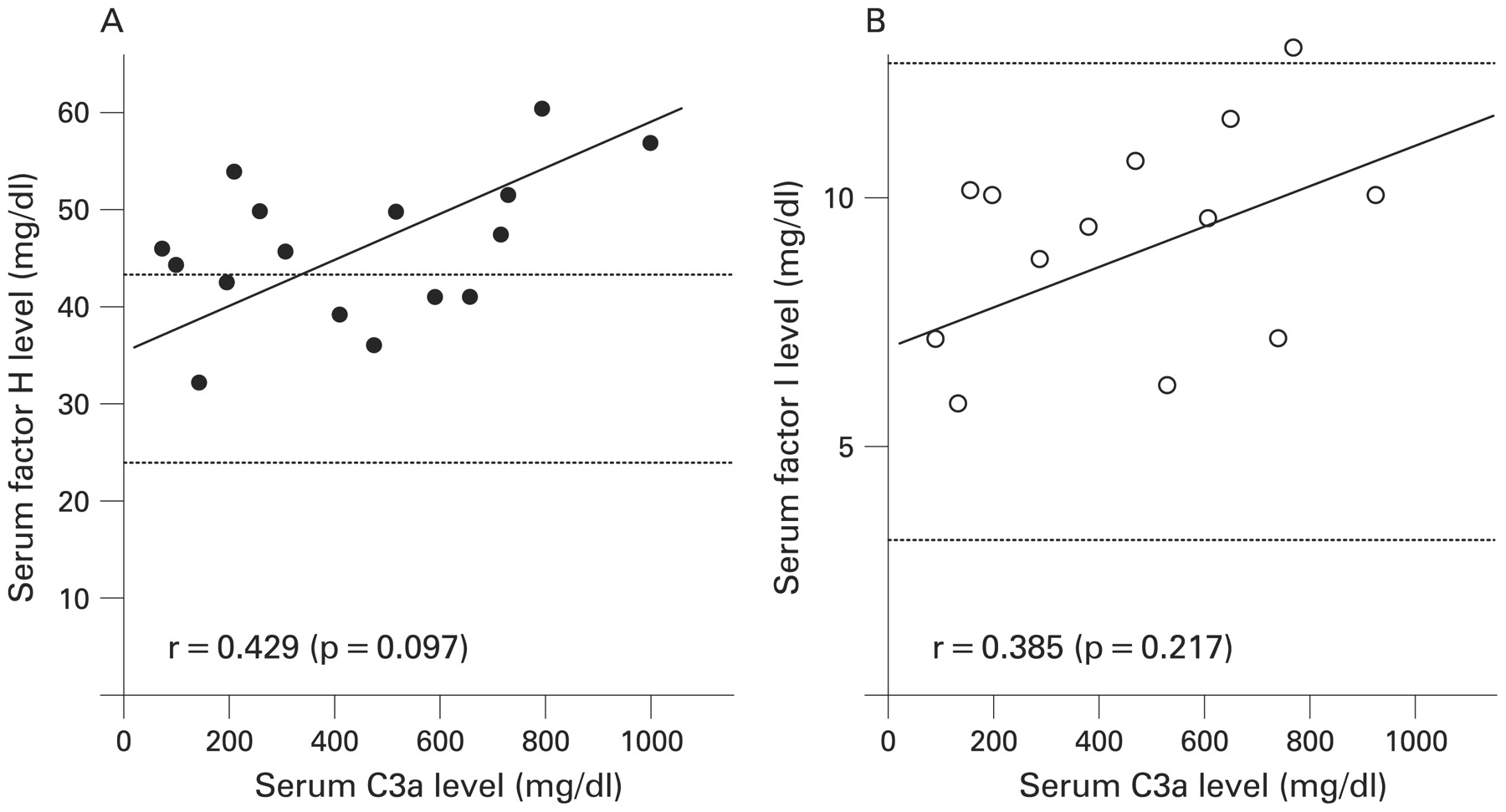
Complement regulatory factor levels in patients with primary antiphospholipid syndrome (APS). Serum factor H (A) and factor I (B) were measured in 16 and 13 patients with primary APS, respectively. Dotted lines represent the upper and lower limits of the normal ranges. Statistical analysis by Pearson correlation coefficient.
Serum immune complex levels
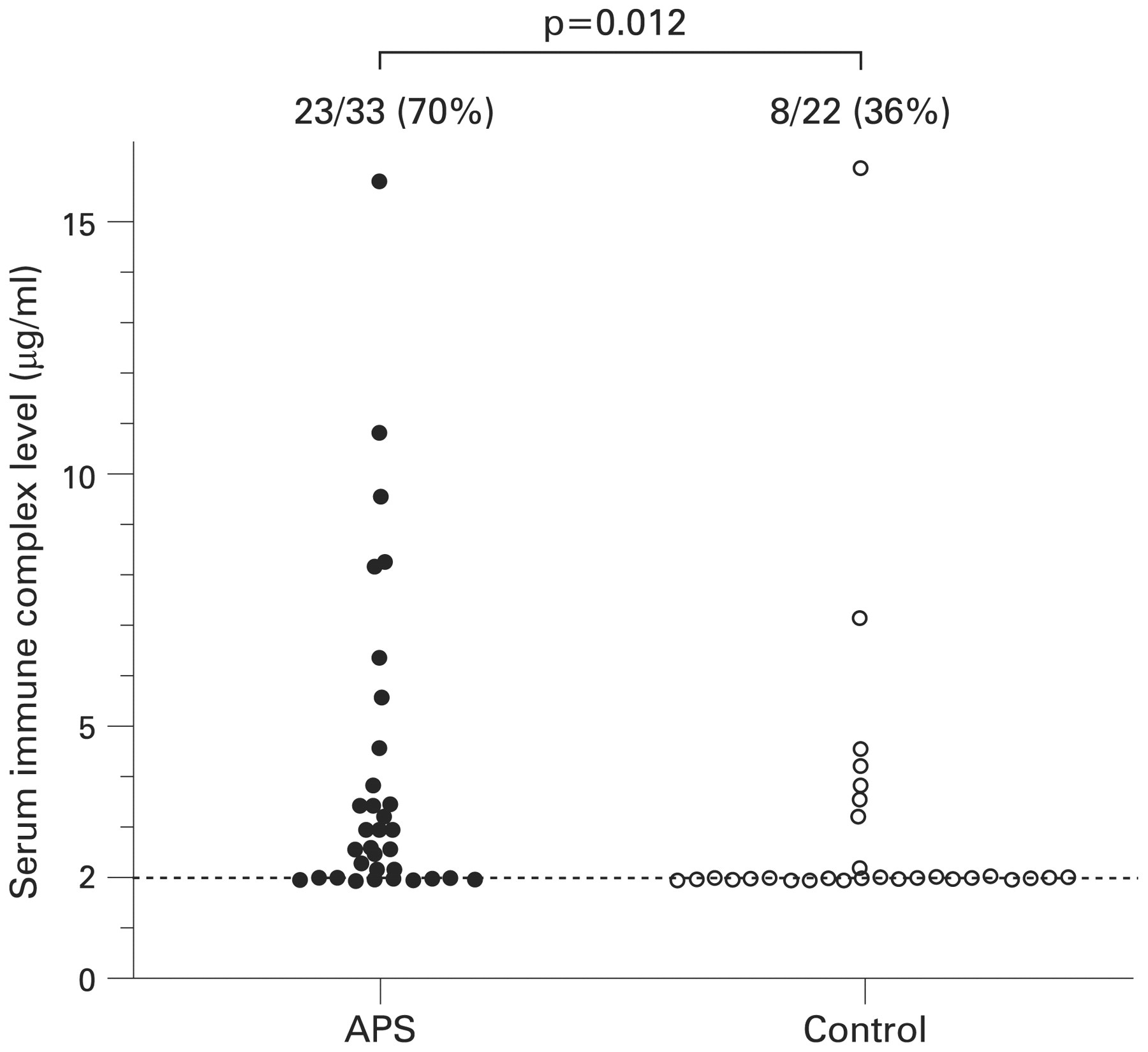
The positive ratio of serum immune complex in patients with primary APS was 23/33 (70%), which was significantly increased compared with ratio of patients with non-SLE connective tissue disease (8/22 (36%), OR = 4.03, 95% CI 1.28 to 12.6) (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Serum immune complex levels. Serum immune complex levels were examined in patients with primary antiphospholipid syndrome (APS) and non-systemic lupus erythematosus (SLE) connective tissue disease controls. Figures at the top of the scatter diagram represent the ratio of the patients with detectable serum immune complex level. The dotted line represents the detection limit (2.0 μg/ml) of serum immune complex level. Control, patients with non-SLE connective tissue diseases. Statistical analysis by Fisher’s exact test.
Correlation between clinical manifestations and hypocomplementaemia
Clinical manifestations of APS occurred as venous thrombosis, arterial thrombosis and pregnancy morbidity in 16/36 (44.4%), 19/36 (52.8%) and 3/27 (11.1%) cases, respectively. No particular manifestation was correlated with hypocomplementaemia. The relative risks for having those manifestation in patients with low CH50 were 1.28 (95% CI 0.34 to 4.82), 1.55 (0.41 to 5.78), 1.88 (0.16 to 22.8), respectively. All patients received warfarin and/or antiplatelet agents, but none received heparin or its derivatives.
Six patients with non-SLE connective tissue diseases had past histories of thrombosis (one venous thrombosis and three arterial thrombosis) and no patients showed hypocomplementaemia.
Correlation between aPL levels and hypocomplementaemia
The prevalences of IgG-aCL, IgM-aCL, IgG-anti-β2GPI, IgM-anti-β2GPI, IgG-aPS/PT, IgM-aPS/PT and LA were 26/36 (72%), 9/36 (25%), 21/36 (58%), 8/36 (22%), 21/36 (58%), 18/36 (50%) and 30/36 (83%), respectively, in patients with primary APS. Neither IgG/M-aCL nor IgG/M-anti-β2GPI was correlated with hypocomplementaemia (low CH50), but the presence of IgG/M-aPS/PT positively correlated with hypocomplementaemia (table 2), which occurred significantly more often in patients with primary APS with high LA than in those with low or negative LA (11/14 (79%) vs 8/22 (36%), OR = 6.42, 95% CI 1.37 to 30.1; table 2).
DISCUSSION
Our results show that hypocomplementaemia is frequently found in patients with primary APS. The high serum C3a and C4a levels and the correlation between serum C3a concentrations and low C3 suggests that hypocomplementaemia in these patients is due to complement activation rather than complement deficiency. None of the patients with primary APS had reduced factor H or factor I levels, indicating that complement activation is not caused by deficiency of these factors but presumably by enhanced immune complex formation. In primary APS, immune complex formation might have a bearing on the anticoagulant potential of aPL, given the positive correlation between strong LA and hypocomplementaemia. This relationship is also related to plasma TNFα released by procoagulant and proinflammatory cells, further supporting a role for complement activation in some manifestations in patients with APS.
Concurrent reduction of C3, C4 and CH50 was the most common profile in our patients, reflecting activation of the classical complement pathway. The proinflammatory effect of C5a is relevant to the pathogenesis of miscarriages in the APS animal model, but none of the patients had raised serum C5a. The finding of lower serum C5a compared with C3a and C4a is consistent with reports in other diseases, including SLE.28–30 C5a is a strong inflammatory mediator and regulatory factors such as factor H or factor I inhibit C3b-dependent activation of C5. In this study, patients with primary APS with high serum C3a levels tended to have raised factor H, and persistent C3 activation may lead to upregulated production of regulatory factors; however, none of the patients were in the acute phase of thrombosis at the time of blood collection. It is likely that the behaviour of C5a in primary APS is similar to that in SLE flares,30 but complement activation is common during pregnancy31 and C5 activation may occur in APS pregnancy. A recent report provides evidence to show that serum of patients with APS can induce tissue factor production on neutrophils and this effect was shown to be due to C5a activation.32 Anaphylatoxins, especially C5a, are extremely labile and are quickly degraded by serum protease, and thus blood was examined immediately; however, it is possible that C5a was degraded in some cases.
Infection, injury or other biological stresses can activate the complement system.33 Accumulating evidence indicates that tissue ischaemia or platelet aggregation can induce complement activation,34 35 therefore, thrombosis itself is one of the incidents which might induce complement activation. However, none of the participants in this study had complications associated with infection, malignancy, impaired circulation, tissue ischaemia, or thrombosis at the time of blood collection and all were negative for C-reactive protein. The hypothesis is further suggested by the data that non-SLE controls who had a history of thrombosis did not show hypocomplementaemia. No patients were receiving heparin or its derivatives, which are known to modify complement activation. Additionally, most of the patients with APS were receiving one or more antiplatelet agents and blood of all patients with primary APS was not examined at the acute stage of thrombosis. In SLE or other immune complex-mediated diseases, immune complex formation can promote activation of the classical pathway. We showed in this study that raised immune complex levels were frequently found in patients with primary APS, suggesting the circulating immune complex potentiates the triggering of complement activation, ultimately leading to thrombotic events. Those immune complexes may include an antigen–antiphospholipid antibody complex in patients with primary APS, as previously described.36 We found that LA activity was correlated with hypocomplementaemia, and LA from APS plasma may reflect the sum of multiple aPL involved in the pathophysiology of APS. Therefore, our results partially support the hypothesis that an aPL–autoantigen complex drives complement activation.
The prevalence of aPS/PT in patients with hypocomplementaemia was higher than that in patients with a normal CH50 level. IgG1 has been proposed as a dominant subclass in aCL in patients with thrombotic events,37 but we7 and others38 have found that IgG2 is the dominant IgG subclass of aCL or anti-β2GPI in APS. Since IgG2 is less effective in activating complement than other subclasses, aPS/PT might be more potent in activating complement than aCL or anti-β2GPI. Additional mechanisms may contribute to complement activation in patients with primary APS. In SLE, reduced complement receptor type 1 levels on erythrocytes and impaired CD55 and CD59 expression (downregulators of the complement system) have been proposed as additional mechanisms of complement activation.39 Equivalent data are unavailable in primary APS, but since many patients with primary APS evolve towards SLE, there may be common mechanisms of complement activation in SLE and APS.
We have previously reported that raised plasma TNFα levels in APS are not associated with HLA haplotypes including class III.40 In this study, some patients with hypocomplementaemia showed raised TNFα. Although the relationship of hypocomplementaemia and plasma TNF levels shown in this study was borderline, as TNFα is produced by activated monocytes, complement activation might accelerate prothrombotic reactions in APS, further supporting a role for complement activation in some manifestations in patients with APS. In the animal APS pregnancy model, TNFα has been proposed as a candidate therapeutic target,41 and our data in patients with APS partly support this idea. Recognition of cross-talk between complement activation and prothrombotic status highlights an important role for the complement system in the physiopathology of primary APS, since complement activation may participate in coagulation processes and contribute to tissue damage. Taken together, these results suggest that the complement system could be a potential therapeutic target in patients with APS.
REFERENCES
Footnotes
Competing interests: None.
Funding: Supported by grants from the Japanese Ministry of Health, Labour, and Welfare and the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Ethics approval: Approval obtained from the local ethics committee.