Article Text

Abstract
Objectives To assess the prevalence of the main causes of morbi-mortality in the antiphospholipid syndrome (APS) during a 10-year-follow-up period and to compare the frequency of early manifestations with those that appeared later.
Methods In 1999, we started an observational study of 1000 APS patients from 13 European countries. All had medical histories documented when entered into the study and were followed prospectively during the ensuing 10 years.
Results 53.1% of the patients had primary APS, 36.2% had APS associated with systemic lupus erythematosus and 10.7% APS associated with other diseases. Thrombotic events appeared in 166 (16.6%) patients during the first 5-year period and in 115 (14.4%) during the second 5-year period. The most common events were strokes, transient ischaemic attacks, deep vein thromboses and pulmonary embolism. 127 (15.5%) women became pregnant (188 pregnancies) and 72.9% of pregnancies succeeded in having one or more live births. The most common obstetric complication was early pregnancy loss (16.5% of the pregnancies). Intrauterine growth restriction (26.3% of the total live births) and prematurity (48.2%) were the most frequent fetal morbidities. 93 (9.3%) patients died and the most frequent causes of death were severe thrombosis (36.5%) and infections (26.9%). Nine (0.9%) cases of catastrophic APS occurred and 5 (55.6%) of them died. The survival probability at 10 years was 90.7%.
Conclusions Patients with APS still develop significant morbidity and mortality despite current treatment. It is imperative to increase the efforts in determining optimal prognostic markers and therapeutic measures to prevent these complications.
- Anticardiolipin Antibodies
- Antiphospholipid Antibodies
- Antiphospholipid Syndrome
- Autoimmune Diseases
- Systemic Lupus Erythematosus
Statistics from Altmetric.com
- Anticardiolipin Antibodies
- Antiphospholipid Antibodies
- Antiphospholipid Syndrome
- Autoimmune Diseases
- Systemic Lupus Erythematosus
Introduction
The antiphospholipid syndrome (APS) is a systemic autoimmune disorder where different prothrombotic factors interact to induce arterial and venous thrombosis.1 It is defined by a combination of at least one clinical feature characterised by vascular thrombosis or pregnancy morbidity and raised titres of antiphospholipid antibodies (aPL), namely lupus anticoagulant (LA), anticardiolipin antibodies (aCL) and/or anti-ß2 glycoprotein-I antibodies.2 ,3 In addition, there is great variety of non-thrombotic and/or non-criteria clinical manifestations related to aPL, but the real prevalence of most of them is unknown.3 ,4 Also, a rare and severe subset of APS has been described (catastrophic APS) that is characterised by multiorgan failure originated by widespread thrombotic disease, which usually affects small vessels over a short period of time and is associated with high mortality rates.5 Furthermore, APS can occur either as a primary condition (primary APS) or in association with other autoimmune diseases, usually systemic lupus erythematosus (SLE), but discordant findings have been reported regarding the relationship of APS and SLE (ie, the percentage of patients with primary APS who develops SLE over time).6 ,7
During the last three decades, several studies of patients with APS have been published, most of them being small or short-term follow-up cohorts.8–10 The Euro-Phospolipid project was created in order to fill some of these gaps in our knowledge of this syndrome. This large international study, composed of 1000 European patients with APS, has helped so far to identify the prevalence and characteristics of the main clinical and immunological manifestations of APS at its onset and during a 5-year follow-up period.3 ,11 ,12
The aims of the present study were to assess the prevalence and characteristics of the main causes of morbidity and mortality after 10-year-follow-up and to compare the frequency of early manifestations with those that appeared later in the evolution of the disease.
Patients and methods
Euro-Phospholipid project
The Euro-Phospholipid project started in 1999 as an attempt to describe the course of APS patients in the real life in Europe. This project was designed as a multicentre, consecutive and prospective observational study. Twenty tertiary university referral centres from 13 European countries (Belgium, Bulgaria, Denmark, France, Germany, Greece, Hungary, Israel, Italy, The Netherlands, Portugal Spain, and the UK) participated in the study with experienced physicians in the management of APS patients. The observation period stopped in 2009 or at the time of the latest patient information if lost to follow-up or at death. The study was performed according to the principles of the Declaration of Helsinki.
Patient selection
In order to gather a sizeable series of patients, 50 consecutive patients from each centre were incorporated into the cohort. The final Euro-Phospholipid cohort included 1000 unselected patients who met the proposed preliminary criteria for the classification of definite APS.2
The patients had been attending the referral centres between the years 1990 and 1999, and since then all patients have been followed up by the same physicians during the ensuing 10 years (1999–2009) with regular visits to the outpatient clinics at least every 3–6 months, depending on the severity of the disease and admitted to hospital if necessary. Specifically, they attended departments of rheumatology (seven centres), clinical immunology/autoimmune diseases (six centres), internal medicine (five centres) and haematology/haemostasis (two centres).
All patients had their medical histories documented and underwent a medical interview as well as a routine general physical examination by a qualified internist and/or rheumatologist. A protocol form was used to record the clinical and serological characteristics of the patients.3 The underlying autoimmune disease was considered when the patient met the specific criteria as previously established.13–18
Definition of clinical features
To minimise possible interobserver bias and to monitor the accuracy of the data collection, a standard protocol was used and the inclusion criteria and variables of this protocol were carefully discussed by all the participating physicians on several occasions. These variables included causes of morbidity (APS manifestations and other associated medical problems), causes of death and survival. The main APS clinical manifestations evaluated in this prospective study have been described previously.3 ,19 ,20 Patients were considered to have catastrophic APS if they presented with an acutely devastating APS with multiple organ (>3) involvement, mainly affecting small vessels supplying organs and presenting over a short period of time (<1 week).21
Pregnancy morbidity was considered when the following established definitions were fulfilled: (a) one or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation, with normal fetal morphology documented by ultrasound or by direct examination of the fetus; or (b) one or more premature births of a morphologically normal neonate before the 34th week of gestation because of eclampsia or severe pre-eclampsia defined according to standard definitions, or recognised features of placental insufficiency; or (c) three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomical or hormonal abnormalities and paternal and maternal chromosomal causes excluded.2
The main clinical manifestations related to SLE evaluated in this study were defined according to the American Rheumatism Association glossary committee and are described in detail elsewhere.22 Diagnoses of vascular risk factors and other associated medical problems that appeared during the study (infections, malignancies, haemorrhages, drug side effects, etc.) were performed on clinical grounds and confirmed by appropriate complementary techniques.11 ,12 The causes of death were based on information obtained from the clinicians in charge, autopsy reports and/or death certificates.
Laboratory studies
The presence of aCL of IgG and IgM isotypes was measured by a β2-glycoprotein I-dependent ELISA.23 They were considered positive if present in medium to high titre (>40 GPL or >40 MPL) on two or more occasions, at least 6 weeks apart. LA activity was detected by coagulation assays according to the guidelines of the International Society on Thrombosis and Haemostasis (Scientific Subcommittee on Lupus Anticoagulants/Phospholipid-Dependent Antibodies).24 Antinuclear antibodies (ANA) were determined by indirect immunofluorescence on mouse liver and HEp-2 cell substrate. Anti-double-stranded DNA (anti-dsDNA) antibodies were determined by Farr's ammonium sulfate precipitation technique, ELISA and indirect immunofluorescence on Crithidia luciliae substrate. Precipitating antibodies to extractable nuclear antigens, including Ro/SSA, La/SSB, U1 small nuclear RNP and Sm, were detected by ELISA and counter immunoelectrophoresis using calf and rabbit thymus and human spleen extracts. Rheumatoid factor was detected by latex and Rose–Waaler tests. All these tests were performed in referral laboratories that adhere to strict quality controls and are participating in the standardisation project of the European Forum on aPL.
Statistical analysis
The following independent variables were included in the statistical analysis for the detection of possible predictors of the outcomes of interest (ie, thrombotic events, pregnancy morbidity and mortality): primary APS, SLE, gender, age at onset of APS, comorbidities (diabetes mellitus, hypertension, dyslipidemia and smoking) and aPL (IgG aCL, IgM aCL and LA). Univariate and multivariate Cox proportional hazard regression tests were used to determine associations of our outcomes of interest (dependent variables) with the different independent variables. When several independent variables appeared to have statistical significance in the univariate analysis, they were included in the multivariate analysis. Results of the analysis of continuous variables are indicated as mean SD. Survival time was defined as the interval from the time the patient entered in the study until death or last contact. Survival probabilities were calculated according to Kaplan–Meier lifetime analysis method. The Standardized Mortality Ratio (SMR) was used to compare the mortality risk of patients with APS to that of the general population of the European Union. SMR and its asymptotic 95% CI were calculated as follows:
where O is the observed number of deaths in the study population and E is the expected number of deaths.25 Statistical significance was defined as a p Value <0.05. Statistical analyses were performed by means of the SPSS program (SPSS, Chicago, Illinois, USA) using the information stored in the database program.
Results
Patient characteristics at the beginning of the prospective study
The entire cohort consisted of 820 (82.0%) female and 180 (18.0%) male patients. There were 985 (98.5%) Caucasians, 5 (0.5%) blacks and 10 (1.0%) patients of other races. Mean (SD) age at study entry was 42 (14) years (range 0–82; median 40). The most prevalent thrombotic risk factors were smoking, hypertension and dyslipidemia, present in 15–20% of the total cohort. In total, 53.1% of the patients had primary APS, 36.2% had APS associated with SLE, 5.0% APS associated with lupus-like syndrome and 5.7% APS associated with other diseases. During the study, eight patients diagnosed at entry as primary APS developed anti-dsDNA antibodies and were reclassified as lupus-like syndrome; also during the last 5 years of the study, three patients were reclassified as SLE-associated APS. During the 10 years of the study, 419 (41.9%) patients were lost to follow-up, excluding deaths (mean 4% every year): 151 patients during the first 5 years and 215 during the ensuing 5 years.
The main clinical manifestations at the onset of the disease, the cumulative clinical manifestations from the onset of the disease until the beginning of the study and the immunological findings when the patients entered into the study and during the initial 5 years of follow-up have been reported in detail elsewhere.3 ,11
APS manifestations during the study period
The frequencies of the main APS clinical manifestations accumulated from the onset of the disease to the inclusion in the study and during the 10-year follow-up are shown in table 1. We compared the manifestations that appeared during the initial 5 years of the observational period (1999–2004) with those that appeared in the ensuing 5 years (2004–2009).
Main clinical manifestations related to the APS that appeared during the 10-year follow-up (1999–2009) of the total cohort of 1000 patients
During the 10-year period, the most frequent manifestation was thrombosis. Thrombotic events appeared in 166 (16.6%) patients during the first 5-year period of the study and in 118 patients (15.3%) during the second period. The most common thrombotic events were strokes (5.3% of the total cohort), transient ischaemic attacks (4.7%), deep vein thromboses (4.3%) and pulmonary embolism (3.5%). Similar proportions appeared in the first and the second periods.
Additionally, other APS-related manifestations not included in the classification criteria for APS (non-criteria APS manifestations) were also commonly observed during the study, including thrombocytopenia (8.7%), livedo reticularis (8.1%), autoimmune haemolytic anaemia (4%), valve thickening/dysfunction (4.6%), epilepsy (3.2%) and skin ulcers (3.1%) (table 1).
During the 10-year follow-up period, 127 (15.5%) women became pregnant (range 1–4). A total of 188 pregnancies occurred and 72.9% of pregnancies succeeded in having one or more live births (range 1–3; total number of live births 137). The most common obstetric complication was early pregnancy loss (<10 weeks) that appeared in 16.5% of pregnancies. The obstetric morbidity rate was lower during the last 5 years of the study so that no women developed pre-eclampsia/eclampsia. Regarding fetal morbidity, the most frequent manifestations were birth prematurity (48.2% of the total live birth) and intrauterine growth restriction (26.3%).
A total of nine patients developed an episode of catastrophic APS during the first 5-year follow-up period but none during the second period.
In the multivariate analysis, no statistical differences were detected in the occurrence of APS manifestations depending on the gender, the age at onset of APS or the presence of comorbidities. The most common underlying autoimmune disease was SLE. Of those 132 (22.7%) patients with APS associated with SLE, 84% were females and 16% males. The mean age (SD) at inclusion was 46 (10) years, and this was lower in comparison with those with primary APS (50 (12) years, p<0.002). Regarding clinical manifestations (SLE vs primary APS), we observed that both groups had similar thrombotic and obstetric profiles, with some exceptions that have significant statistical differences. Patients with APS associated with SLE developed more frequently arthralgias (31.1% vs 8.1%) and arthritis (21.2% vs 2.8%), leucopenia (14.4% vs 2.4%), autoimmune haemolytic anaemia (15.9% vs 2.1%), livedo reticularis (21.2% vs 6.9%), epilepsy (6.8% vs 1.2%), glomerular thrombosis (3.0% vs 0.2%) and myocardial infarction (3.8% vs 1.2%) compared with primary APS patients who developed more frequently superficial thrombophlebitis (0% vs 1.9%) and fetal morbidity such as birth prematurity (40% vs 72.3%) and intrauterine growth restriction (1% vs 51.1%). Tables 2 and 3 depict the main thrombotic and obstetric manifestations related to APS associated with SLE and primary APS patients.
Main thrombotic manifestations related to APS associated with SLE and primary APS that appeared during the 10-year follow-up (1999–2009) of the ‘Euro-Phospholipid’ cohort
Main obstetric manifestations related to APS associated with SLE and primary APS that appeared during the 10-year follow-up (1999–2009) of the ‘Euro-Phospholipid’ cohort
Laboratory features during the study period
The most prevalent immunological features during the 10-year follow-up period were aCL (81.7%), LA (36.3%), ANA (36.3%) and anti-dsDNA antibodies (12.7%). No significant statistical differences were found in the clinical presentation of the APS according to the presence or absence of any of these antibodies. Furthermore, LA, ANA, anti-dsDNA, anti-Ro/SSA, anti-La/SSB, anti-RNP and anti-Sm antibodies were more frequent in patients with APS associated with SLE compared with primary APS (p<0.001).
APS treatment during the study period
During the 10-year study, the majority of patients received antithrombotic treatment. Low doses of antiaggregant agents were used in 35.0% of the patients during the initial 5-year period and in 27.7% of the patients during the ensuing 5 years (86% aspirin and 12% other antiplatelet). In this last period, double antiplatelet therapy was administered in 1.8% of patients. On the other hand, 40.2% of the patients received oral anticoagulants during the first period and 37.0% of patients during the second follow-up period (62% of them at a target international normalised ratio (INR) of 2–3 and 38% at a target INR 3–4). Also during this last 5 years, 12.4% of patients were taking the association of antiplatelet and anticoagulant treatment and 21.1% of patients did not receive any kind of antithrombotic treatment.
During the last 5-year follow-up period, 24.8% of patients on antithrombotic treatment (20% antiplatelet, 45% anticoagulant, 35% both treatments associated) and 5.2% of patients without any antithrombotic treatment developed thrombosis. In addition, 8.3% of patients under antithrombotic therapy (62.2% antiaggregant, 29.7% anticoagulant, 8.1% both treatments associated) and 1.5% of patients without any antithrombotic treatment developed APS-related obstetric complications.
Of those 135 patients who did not receive any antithrombotic treatment throughout this follow-up period, 93.3% and 97.0% were free of thrombotic complication and obstetric events, respectively.
No significant statistical differences were found in the antithrombotic treatment related to the underlying autoimmune disease (APS associated with SLE/primary APS).
During the 10-year study, 61 major bleeding episodes were developed in patients on antithrombotic treatments, 33% of them at a target INR>3 (50.8% musculocutaneous, 24.6% cerebral, 16.4% gastrointestinal and 8.2% intra-abdominal) of which 10 (16.4%) were fatal.
Mortality and causes of death during the study period
During the 10-year period, 93 (9.3%) patients (72 female and 21 male) died. The mortality rate was similar in both periods: 5.3% in the initial 5-year follow-up period and 4% in the ensuing 5 years. Mean (SD) age at death was 59 (14) years (range 19–94). The unadjusted SMR for the total cohort was 1.8 (95% CI 1.5 to 2.1) in comparison with the general population of the area studied. There was no difference in the mortality rate regarding underlying disease: 6.8% of patients with APS associated with SLE and 7.1% of patients with primary APS died.
During the observational period, after performing the multivariate analysis, no statistical differences were detected in the causes of death depending on the underlying disease (primary APS or SLE) or on the treatment that patients were receiving (immunosuppressive or anticoagulant agents).
The most common causes of death were severe thrombotic events, including myocardial infarction, strokes and pulmonary embolism (36.5% of total deaths), infections (26.9%: bacterial 21.5%, other 5.4%) and haemorrhages (10.7%). Five out of the nine (55.6%) patients who developed catastrophic APS died. Table 4 shows the main causes of death of the total cohort and table 5 related to primary APS and APS associated with SLE.
Causes of death during the 10 year follow-up (1999–2009) of the total cohort of 1000 patients
Main causes of death related to antiphospholipid syndrome (APS) associated with systemic lupus erythematosus (SLE) and primary APS that appeared during the 10-year follow-up (1999–2009) of the ‘Euro-Phospholipid’ cohort
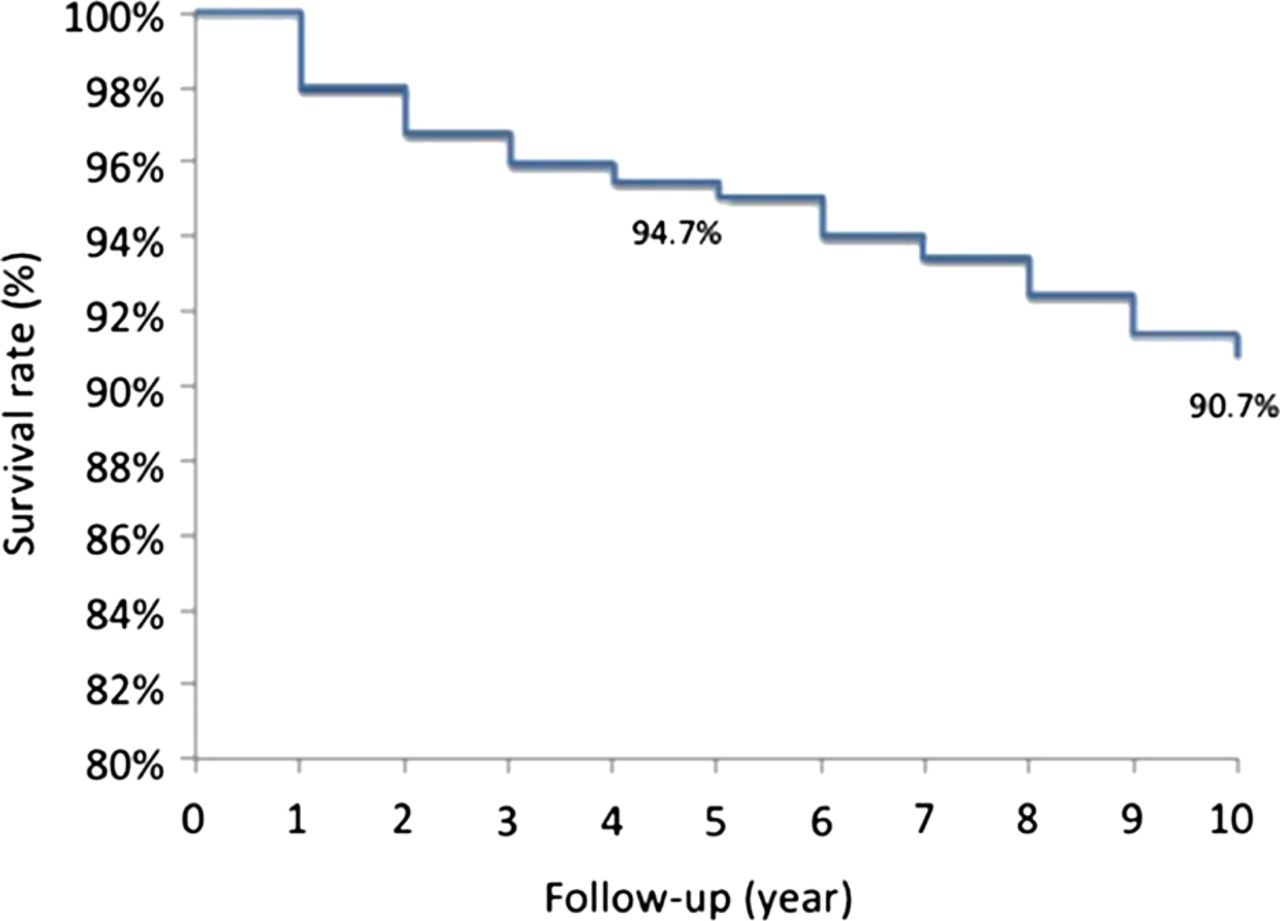
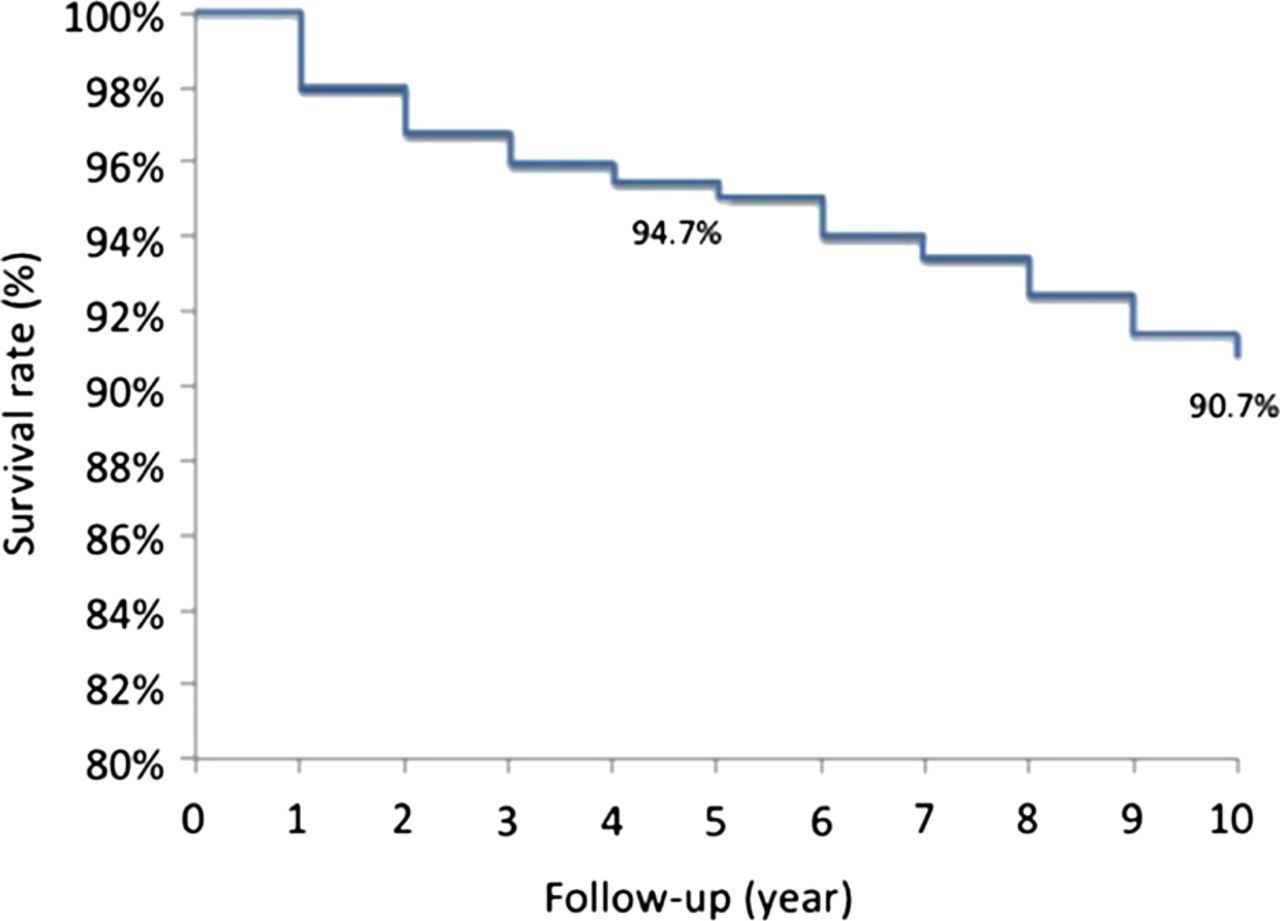
A survival probability of 94% was found at 5 years from the time of entry into the study and of 91% at 10-year follow-up (figure 1). There were no differences in survival probabilities between patients with primary APS and those associated with SLE.
{kind=link}
Kaplan–Meier survival curve of the total cohort showing a 94.7% probability of remaining alive at 5 years and 90.7% at 10 years from the time of entry into the study.
Discussion
The Euro-Phospholipid project is a prospective observational study of a cohort of 1000 European patients that fulfilled classification criteria of definite APS.2 In this paper, we have described the prevalence and characteristic of the main clinical manifestations as well as the mortality rate and causes of death in this cohort during a 10-year period (1999–2009). In addition, we compared these results with the accumulated data from the onset of the disease to the inclusion in this study.3
The strength of this study, as compared with other European series,10 ,26 ,27 lies in its multicentre design, including centres from countries across all Europe, thus covering a very representative sample of the European APS population. The limitation regarding the possible differences between the participating hospitals has been overcome by a strict selection of tertiary referral university centres with physicians experienced in the management of APS and by establishing a well-defined protocol. A potential selection bias by solely choosing tertiary referral centres is improbable because the majority of patients with APS from these countries were referred to these centres.
Probably, the major study limitation has been the losses during the 10-year period of follow-up (41.9% of patients, excluding deaths), but this accounted for only 4% of the total cohort every year and appearance or absence of the different outcome variables during the period of time that these patients participated in the study was also registered. Therefore, this cohort can be considered representative of what are currently accepted to be APS patients in Europe.
In this European cohort, the most common clinical manifestations were thrombotic events, with a similar incidence among the first 5 years of follow-up period compared with the ensuing 5 years. However, the frequencies are lower compared with the cumulative clinical manifestations before the start of the study (median previous period of evolution, 6 years)3 and compared with other studies in other European,10 American28 and Asian series.29 These lower frequencies could be due to genetic or environmental differences, but could also reflect the effect of medical care during the study because of the prospective nature of the Euro-Phospholipid project. Interestingly, the most common thrombotic events at baseline were deep vein thromboses, but during the study we observed a higher incidence of arterial thrombotic events (stroke, transient ischaemic attacks and acute myocardial infarction), as it has been also found in other studies.28 ,30
Additionally, several APS-related manifestations not included in the classification criteria were also common, including haematological manifestations (thrombocytopenia and haemolytic anaemia), livedo reticularis, epilepsy, skin ulcers and valve thickening/dysfunction.
One of the characteristic clinical manifestations of the APS is fetal morbidity, including early and late pregnancy losses, intrauterine growth restriction and prematurity. Additionally, maternal morbidity (pre-eclampsia, eclampsia and abruptio placentae) is also relatively common in pregnant patients with APS. The most common fetal complications in our cohort were premature birth and intrauterine growth restriction, while the most common obstetric maternal complication was pre-eclampsia. During the study, only a small percentage of patients from this cohort became pregnant (15.5%) compared with a higher proportion of patients with history of pregnancy at the study entry (71.9%).3 However, 72.9% succeeded in having one or more live births being significantly higher than the 47.6% at baseline. Probably because of this increase in successful pregnancies, the percentage of fetal morbidity (intrauterine growth restriction and prematurity births) was higher.3 Also, pregnancy loss and obstetric morbidity (pre-eclampsia/eclampsia) rates were lower during the study and were reduced to more than half in the last 5 years of follow-up. These lower frequencies probably reflect the effect of therapy and medical care during the study, but also represent the improvement in the management of the obstetric APS patients during the last decade with the widespread use of antiaggregant and anticoagulant drugs and the careful monitoring of pregnancies.31–33
The catastrophic APS was described in 1992 by Asherson34 as a APS subset characterised by multiple vascular occlusive events, usually affecting small vessels supplying organs and presenting over a short period of time (less than a week). It is considered to be one of the most severe complications of the APS, and about half of the patients succumb despite seemingly adequate treatment, including anticoagulation, steroids, plasma exchange and intravenous immunoglobulins.35 In this study, the incidence of this catastrophic subset in the 10-year period was found to be around 0.9% and the mortality rate was 55.6% (5/9 patients), similar to other patient series in the same decade.36
The frequencies of the major immunological APS features in our cohort were also comparable to those of other reports.3 ,6 ,37 Neither individual aPL nor the combination of some of them was associated with an increased incidence of any specific clinical manifestation. This lack of association during the study period is probably due to the relatively low prevalence of new APS clinical manifestations. In addition to aPL, ANA were detected at some time during the course of the illness, but usually at low titres, and high titres of anti-dsDNA antibodies were found in patients with associated SLE. As in the baseline study, we observed that patients with APS associated with SLE had more episodes of arthritis, leucopenia, anaemia, livedo reticularis, epilepsy and glomerular thrombosis compared with primary APS.3
The vast majority of patients in this cohort were under antithrombotic treatment during the 10-year follow-up period. While treatment has led to a reduction in the frequency of venous thrombotic events compared with baseline data, it does not seem to produce an important impact on the frequency of arterial events. This may be related to the fact that this particular group of patients may need a higher anticoagulation target (INR 3–4) as well as strict control of comorbidities and prothrombotic factors. Moreover, some authors had suggested that the concomitant use of immunosuppressive drugs such as those that produce B cell modulation/depletion or the use of antimalarials might help prevent the recurrence of new arterial events.38
On the other hand, 23% of patients in this cohort were not receiving antithrombotic treatment during this study period. This may reflect the ‘real world’ situation of this disease. Many physicians may have discontinued anticoagulant treatment after long-term follow-up without recurrent events, while others may still be unenthusiastic to prescribe primary thromboprophylaxis to those female patients with APS who had experienced pregnancy morbidity or long-term secondary thromboprophylaxis several years after the APS thrombotic event.
Despite the overall reduction of thrombotic events during the 10 years of follow-up as compared with baseline data, low-dose aspirin did not prevent thrombotic events in 79 patients, 90 patients on anticoagulants presented new recurrent thrombotic events during the initial 5-year period (21 of them at a target INR >3) and 87 patients during the ensuing 5-year period (37 of them at a target INR >3). This may reflect a selection bias (patients with more severe clinical manifestations may be more consistently treated with oral anticoagulants by their doctors), but we believe that this was probably because many patients with APS could not keep their target INR. Therefore, the INR at the time of the event would have much more value than the target INR, but these data were not consistently obtained in the study. Also, this difficulty to obtain a stable INR may be a reason to develop some adverse effects of treatment such as bleeding. Sixty-one haemorrhages occurred during the 10-year follow-up period; 30 of them in internal organs and 10 of them were the main cause of death. These problems of current oral vitamin K antagonists having numerous drug and dietary interactions that lead to an obliged regular, inconvenient and costly monitoring of the INR may be overpassed by the use of novel anticoagulants (competitive and reversible direct factor IIa and Xa inhibitors). However, adequately powered prospective randomised controlled multicentre clinical trials are needed to compare the efficacy of the new oral anticoagulants versus oral vitamin K antagonists in patients with thrombotic APS.
The total mortality rate during the observational period was 9.3%, higher than the mortality rate in a European SLE cohort of 1000 patients (‘Euro-lupus cohort’) (6.8%) during a similar period.39 The predominant causes of death were severe thromboses (stroke, myocardial infarction, pulmonary embolism or catastrophic APS) and accounted for more than one-third of deaths. In the ‘Euro-lupus cohort’, all patients who died because of thrombosis had SLE associated with APS, and the most common fatal thrombosis were the same and with similar incidences rates to that in the Euro-Phospholipid project.39
When we compared the initial 5 years of follow-up with the ensuing 5 years, mortality rates were similar in both follow-up periods. However, some differences were detected: the fatal thrombotic events were higher during the initial period and the incidence of death because of malignancy becoming more frequent during the last period. Also, infections and bleedings accounted for one-third of deaths during the long-term study, appearing in similar percentages during both periods.
We were not able to identify any clinical or immunological parameter with prognostic significance for mortality. Interestingly, although survival probability decreased during the 10-year period, in our study older age was not associated with increased mortality. This may be due to the fact the APS-related conditions dominated the causes of mortality in this cohort, but also because the population was not very old.
In conclusion, this long-term study provides updated information on APS morbidity and mortality characteristics. Patients with APS still develop significant morbidity and mortality despite current treatment (mainly oral anticoagulants and/or antiaggregant agents); therefore, it is imperative to increase the effort in determining optimal prognostic markers and therapeutic measures to prevent these important complications of the APS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Collaborators See complete list of members of the Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies) in online supplementary appendix A.
Contributors All the coauthors have participated in the design of the study, the collection and analysis of data and writing of the manuscript.
Competing interests None.
Ethics approval Ethics Committee from the Hospital Clínic of Barcelona.
Provenance and peer review Not commissioned; externally peer reviewed.