Article Text

Abstract
Objective To compare the efficacy and safety of enteric-coated mycophenolate sodium (EC-MPS) versus azathioprine (AZA) in patients with active systemic lupus erythematosus (SLE) disease.
Methods A multicentre, 24-month, superiority, open-label, randomised controlled trial (NCT01112215) was conducted with 240 patients (120 per arm) receiving either EC-MPS (target dose: 1440 mg/day) or AZA (target dose: 2 mg/kg/day) in addition to prednisone and/or antimalarials. The primary endpoint was the proportion of patients achieving clinical remission, assessed by SLE Disease Activity Index 2000 (SLEDAI-2K) and British Isles Lupus Assessment Group (BILAG), at 3 and 24 months. Secondary endpoints included time to clinical remission, BILAG A and B flare rates, time to flare, corticosteroid reduction and adverse events (AEs).
Results Proportion of patients achieving clinical remission (clinical SLEDAI=0) was higher in the EC-MPS group at 3 (32.5% vs 19.2%; treatment difference, 13.3 (CI 2.3 to 24), p=0.034) and 24 months (71.2% vs 48.3%; treatment difference, 22.9 (CI 10.4 to 34.4), p<0.001). EC-MPS was superior with respect to time to clinical remission (HR 1.43; 95% CI 1.07 to 1.91; p=0.017). BILAG A/B and B flares occurred more frequently in the AZA group (71.7% vs 50%, p=0.001 and 21.67% vs 8.3%, p=0.004, respectively). EC-MPS was superior with respect to time to first BILAG A/B (HR 1.81; 95% CI 1.3 to 2.56; p=0.0004) and BILAG A flare (HR 2.84; 95% CI 1.37 to 5.89; p=0.003). AEs were similar in both groups except for leucopenia that occurred more frequently with AZA.
Conclusions EC-MPS was superior to AZA in treating SLE and preventing further relapses.
Trial registration number NCT01112215; Results.
- Systemic Lupus Erythematosus
- Treatment
- Outcomes research
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune disease characterised by heterogeneous clinical manifestations and a relapsing–remitting course. Although there have been significant improvements in its prognosis and management, the treatment of moderate-to-severe SLE remains unsatisfactory with a significant proportion of patients still having morbidity, poorer quality of life and increased mortality.1
Controlled clinical trials in SLE have focused primarily on lupus nephritis (LN) and generally have not analysed non-renal manifestations.2–4 Standard initial therapy for extrarenal disease consists of oral corticosteroids and antimalarials, although immunosuppressive drugs are needed to control disease activity, minimise SLE organ damage and reduce corticosteroids. To date, data on the efficacy, safety and steroid-sparing effects of non-biological therapies are limited and provided mainly by small open-label studies and few randomised controlled trials (RCTs).5–11 Historically, azathioprine (AZA) has been one of the most frequently used immunosuppressants with the advantage of its safety during pregnancy. There is modest evidence supporting its use, and side effects sufficient to discontinue the drug have been described in about one-third of cases.10 However, evidence suggests these might be mitigated by measuring 6-thioguanine (6-TGN) levels.7 8 10 12 Results from the RCTs conducted in non-renal SLE have shown that low-dose ciclosporin is as effective as AZA in severe SLE as a steroid-sparing agent,10 and that leflunomide9 and methotrexate5 are more effective than placebo in mild-to-moderate active disease.
Mycophenolate mofetil (MMF) and enteric-coated mycophenolate sodium (EC-MPS) have become widely used for SLE, although most data come from LN studies using MMF. Initial RCTs have shown MMF to be at least as effective as cyclophosphamide (CYC) for induction therapy and equal or superior to AZA in maintaining renal response.4 13–15 EC-MPS has been shown to have similar efficacy to MMF but with fewer gastrointestinal side effects16 and has been increasingly used for adult and paediatric patients.17 18 To date, there have been no robust trials assessing the non-renal effects of these agents. However, limited data from open-label studies and underpowered RCTs17–24 in refractory SLE have shown MMF to be as effective as CYC for ameliorating non-renal symptoms in patients with LN,23 comparatively better in dermatological and haematological manifestations19 and to reduce disease activity and act as a steroid-sparing agent.21
To confirm the relative efficacy and safety of EC-MPS to AZA for active non-renal lupus disease, we conducted this 24-month clinical trial.
Methods
The study was conducted at 12 teaching hospitals in Spain between May 2010 and January 2016 in accordance with the Declaration of Helsinki and Good Clinical Practice principles. All participants provided written informed consent. The study protocol was reviewed and approved by every participant centre.
Patient eligibility and enrolment
Eligible patients were aged ≥18 years, had an SLE according to the revised ACR classification criteria25 and moderate-to-severe active disease defined as: a SLE Disease Activity Index 2000 (SLEDAI-2K)26 total score ≥6 or at least 1 British Isles Lupus Assessment Group (BILAG) A or 2 BILAG B domain scores at screening.27 Key exclusion criteria were immunosuppressant therapy 12 weeks before randomisation; active nephritis or non-lupus-related significant laboratory abnormalities. See online supplementary material for detailed inclusion and exclusion criteria.
Randomisation
The randomised list, stratified by centre and SLEDAI-2K score (6–9 vs ≥10), was created using computer-generated random-number sequences in blocks of 10 (C4 Study Design Pack Software, GlaxoSmithKline) by the Vall d’Hebrón Hospital investigational pharmacist, who was blind to patient enrolment. Sequentially numbered, concealed envelopes containing group assignment were provided to the investigators.
Eligible patients were randomised (1:1) to receive EC-MPS (target dose: 1440 mg/day) or AZA (target dose: 2 mg/kg, per thiopurine methyltransferase levels (TPMT)) in addition to background oral prednisone and antimalarial agents. Patients unable to tolerate the target dose or whose weight was below 50 kg remained in the study if they tolerated a minimum daily dose of either 720 mg of EC-MPS or 50 mg of AZA during the first 6 months. Progressive immunosuppressant dose reduction was allowed after week 24 on a 3- to 6-monthly basis per clinical judgement. Changes in antimalarial and prednisone doses were not restricted (see online supplementary text).
Outcomes and follow-up
The primary efficacy endpoints were the proportion of patients achieving at 3 and 24 months, at least 8 consecutive weeks of clinical remission (CR), defined as a clinical SLEDAI-2K=0, where serology was permitted (maximum SLEDAI=4) following the later Zen et al 28 29 equivalent definition, in the absence of any BILAG A, B or C score.
Secondary endpoints included: the overall proportion of patients in CR and partial clinical response (PR) (≥50% reduction in the total SLEDAI-2K score with a BILAG C score or better, without new BILAG A/B scores); treatment failure (premature discontinuation necessitated by protocol-prohibited rescue therapy due to worsening or persistent disease activity (see online supplementary text)); time to CR; rates of SLE flares, defined as a new BILAG A (severe flare) or B (moderate flare) score in any organ system following a BILAG C, D or E score30; time to flare; changes in total mean SLEDAI-2K and BILAG-2004 scores, prednisone dose and serological activity (anti-double-stranded DNA (dsDNA) antibodies and C3). Comparative SLE Responder Index (SRI)4 31 and Lupus Low Disease Activity State (LLDAS)32 measurements were added post-hoc. The outcomes were adjudicated by an independent assessor.
Patients were evaluated monthly for the first 6 months and every 3 months thereafter. At each visit, the SLEDAI-2K26 and BILAG-200430 33 scores, physician’s global assessment (PGA), changes in concomitant medications and adverse events (AEs) were recorded. To assess the BILAG index global response, the scores were converted to numeric values (A=12, B=3, C=1, D=0, E=0).34 SLICC Damage Index (SDI)35 was scored at baseline and month 24. Patients were followed up for 24 months, regardless of outcome.
Safety assessments included the incidence and severity of AEs classified using the Medical Dictionary for Regulatory Activities (MedDRA version 12) (online at http://www.meddra.org/). SLE flares were not considered AEs.
Statistical analysis
The primary efficacy analysis was an intention-to-treat analysis that included all randomised patients who received at least one dose of the study agents, had at least one measurement prior to administration and had at least one efficacy assessment. The safety population comprised all patients who received at least one dose of study medication. Continuous variables are presented as mean and SD, and categorical variables as count and percentage. The main comparisons of proportions, at 3 and 24 months, have been estimated using generalised estimating equations to account for the repeated measurement design. To adjust for multiple comparisons (3 and 24 months), a Bonferroni correction of the significance level was applied. Continuous data comparisons were performed with t-test. In the time-to-CR analysis, the proportional hazards (PH) assumption of the Cox model regression did not hold. However, the curves clearly did not cross throughout the study. Thus, to provide an easier interpretation, the results are presented as HRs, which may be understood as an ‘average effect’. The time to flare analyses did not show any issue with the PH assumption. Subgroup analyses of the primary endpoint by selected baseline characteristics are presented. Additional comparisons were carried out with log-rank tests. Statistical analyses were performed using SAS software V.9.3. Differences were determined to be statistically significant when two-sided p value was less than 0.05.
The sample size estimation was based on an overall remission rate in AZA-treated patients at 24 months of 45%.6 7 10 Under this assumption, 120 patients would need to be assigned to each group to have 80% power to detect significant differences with a bilateral alpha level of 0.05, assuming a 20% difference between treatments and allowing a 20% dropout rate.
Results
Patients
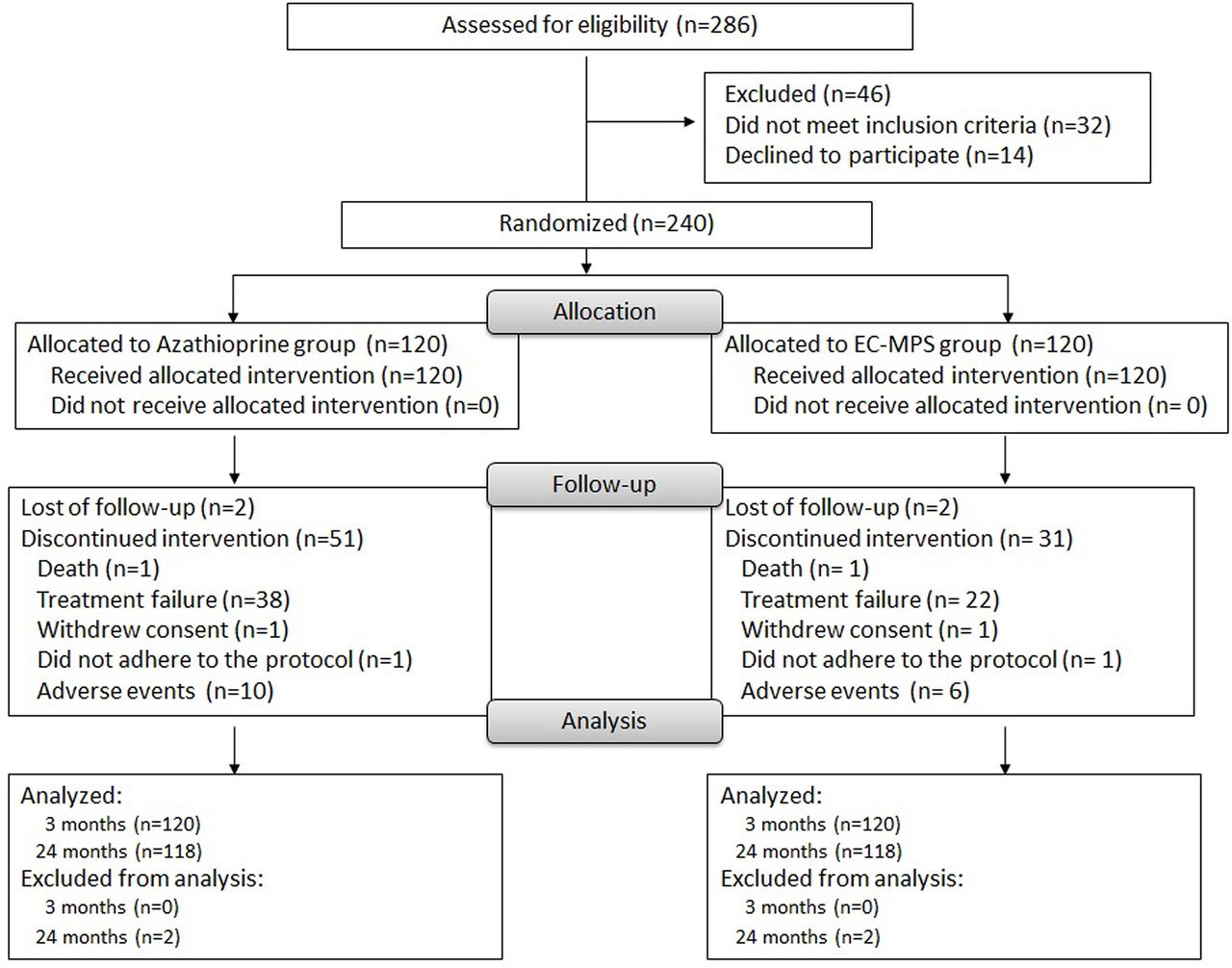
A total of 240 patients were enrolled between May 2010 and December 2013. Of the patients in this intention-to-treat population, 120 were randomised to each treatment group. Baseline demographics are shown in table 1. Seven patients had neurological manifestations: peripheral polyneuropathy1 and transverse myelitis2 in the AZA group; and organic brain syndrome,2 lupus-related Parkinsonism1 and Guillain-Barré1 in the EC-MPS group. A total of 154 patients (64.2%) completed the study: 87 (72.5%) in the EC-MPS group and 67 (55.8%) in the AZA group. Fifty-three patients in the AZA group and 33 in the EC-MPS group discontinued the study. The main reasons for early withdrawal were treatment failure and AEs (figure 1).

Study flow diagram.
Baseline demographic and disease characteristics of patients
Treatment
Mean (SD) doses of EC-MPS and AZA during the study were 1.18 (0.29) g and 123.2 (22) mg, respectively. The proportion of patients whose average daily dose was 80% or more of the target dose at 3 months was 79.2% for EC-MPS and 75.8% for AZA. Mean doses at withdrawal for patients with treatment failure were 1.35 (0.223) g and 133 (20.9) mg, respectively. The mean treatment duration was 575 (min 30 and max 730) days for EC-MPS and 496 (30–730) for AZA. Few patients in CR could discontinue the study agents, 4 (5.8%) in the AZA group and 10 (11.4%) in the EC-MPS group. Antimalarials were discontinued in eight patients (6.7%) in the EC-MPS and in two (1.7%) in the AZA group (see online supplementary table S1).
Outcomes
Primary endpoint
Clinical remission rates were higher in the EC-MPS group by month 3 (32.5% (39/120 patients)) compared with the AZA group (19.2% (23/120); percentage difference 13.3% (95% CI 2.3 to 24), p=0.034) and sustained throughout the study to month 24 (71.2% (84/118) vs 48.3% (57/118); percentage difference 22.9% (95% CI 10.4 to 34.4), p<0.001) (figure 2A, online supplementary table S2). Median time to CR was 6 months (95% CI 5 to 9) in the EC-MPS group and 12 months (95% CI 9 to 16) in the AZA group (p=0.002). The HR for time to CR with EC-MPS use was 1.42 (95% CI 1.07 to 1.90; p=0.017) (figure 2B). There were also more SRI4 and LLDAS responders at 3 months (p=0.053) and at 24 months (p<0.0001) in the EC-MPS group (see online supplementary table S2).

Results for primary efficacy endpoints. Rates of clinical remission during the 24-month study period (A). Cumulative probability for time to clinical remission (B). Mean BILAG index (C) and SLEDAI-2K (D) global scores during the study. Bars in (C) and (D) represent the SEM. AZA, azathioprine; BILAG, British Isles Lupus Assessment Group; EC-MPS, enteric-coated mycophenolate sodium; SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity Index 2000.
SLEDAI-2K and BILAG-2004 scores showed an improvement over time reflecting the reduction in disease activity in both groups. This reduction was superior in the EC-MPS group. BILAG score difference was already statistically significant at month 3 (p=0.011) (figure 2C) whereas SLEDAI-2K score reached statistical significance at month 24 (p=0.006) (figure 2D). Resolution of disease activity (from BILAG A/B to BILAG D) in most individual body systems was similar in both groups, except for the cardiorespiratory domain with more EC-MPS-treated patients reaching CR at 3 months (p=0.015) (see online supplementary figure S1). Subgroup analysis did not show evidence of different clinical response (figure 3). Cumulative rates of treatment failure at 24 months were higher in the AZA group (31.7% (38/120 patients)) compared with the EC-MPS group (18.3% (22/120)) (p=0.099)).

Risk of clinical remission in patient subgroups. The HR was derived from a Cox model, with treatment as the only factor, according to subgroup. AZA, azathioprine; dsDNA, double-stranded DNA; EC-MPS, enteric-coated mycophenolate sodium.
Secondary endpoints
SLE flares
BILAG A/B flares were more common in the AZA group (71.7% (86/120 patients)) compared with the EC-MPS group (50% (60/120)) (p<0.001). In the AZA and EC-MPS groups, 34.2% and 35% patients had 1 disease flare; 21.7% and 13.3% had 2 flares; and 16.7% and 5% had >2 flares, respectively. Mucocutaneous and renal flares were more frequent in the AZA group (p=0.003 and p=0.031, respectively) (figure 4A). Flares were associated with medication reduction in 38 patients (31.7%) of the AZA group and 29 (24.2%) of the EC-MPS group. Rates of new BILAG A flares were low, but significantly higher in AZA (21.7% (26/120) vs 8.3% EC-MPS (10/120), p=0.004) (figure 4B). BILAG A biopsy-proven glomerulonephritis occurred in 5.8% (7/120; all type III–IV) of patients in the AZA group compared with 0.8% (1/120; type V) in the EC-MPS group (p=0.031). The HR for time to first BILAG A/B and BILAG A was 1.84 (95% CI 1.32 to 2.57; p<0.001) and 2.81 (95% CI 1.36 to 5.84; p=0.003), respectively (figure 4C,D). No association with anti-dsDNA antibody positivity or complement levels was found.

{kind=link}
{kind=link}
![[SP1.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1575/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
Results for secondary efficacy endpoints. Proportion of patients with a new flare of SLE over the 24-month study period. Flare is determined by a BILAG index A or B (A). Flare is determined by a BILAG score of A only (B). Cumulative probability of being free of BILAG A/B flare (C) and BILAG A only flare (D). Percentage of patients with corticosteroid dose reduced to ≤7.5 mg/day from ≥7.5 mg/day at baseline (n= 103 in the AZA group and n=98 in the EC-MPS group) (E), and percentage with increased corticosteroid use over 24 months (F). Analyses are based on the intention-to-treat population. Values at the top of the bars in (A) and (B) are actual percentages, with SE represented. AZA, azathioprine; BILAG, British Isles Lupus Assessment Group; EC-MPS, enteric-coated mycophenolate sodium; SLE, systemic lupus erythematosus.
Corticosteroid use
Reduction of the prednisone dose (<7.5) by month 24 among those patients taking ≥7.5 mg/day at inclusion was higher in the EC-MPS group (94.9% (93/98) patients) compared with the AZA group (83.5% (86/103), p=0.027) (figure 4E). During the study, mean prednisone dose decreased from 28.6 (21.2) to 4.2 (2.3) mg/day in the EC-MPS group compared with 23.9 (18.1) to 6.8 (9.2) mg/day in the AZA group (p=0.037). Fewer rescue increments (≥7.5 mg/day) were required with EC-MPS (figure 4F) (see online supplementary table S1). Prednisone discontinuation occurred in 10.5% (12/114) and 17.2% (20/116) of the AZA and EC-MPS groups, respectively.
Changes in immunological parameters
Mean anti-dsDNA antibody level reductions from baseline were greater in the EC-MPS group at month 3 (p<0.001). No differences in mean C3 level increments were observed (see online supplementary figure S2).
![[SP2.jpg]](https://ard.bmj.com/content/annrheumdis/76/9/1575/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
Adverse events
The incidence of AEs was similar in both groups: 59.2% (71/120) patients given EC-MPS and 57.5% (69/120) patients given AZA (p=0.793) (table 2). The rate of serious events was also similar in both groups. Infections were the most common AEs with an overall rate of 32.5% (39/120 patients) in EC-MPS and 27.5% (33/120) in the AZA group (p=0.398). The rate of serious infections was low in both groups: 4.2% (5/120) patients in the EC-MPS group and 5.8% (7/120) in the AZA group.
Incidence of AEs that emerged during treatment and serious AEs
The proportion of patients with AEs leading to withdrawal was slightly higher with AZA (8.3% (10/120)) than with EC-MPS (2.5% (4/120), p=0.06). Leucopenia was more frequent in the AZA group. One death occurred in each group, both due to complicated pneumonia. Three cases of cancer (two breast cancers and one thymoma) occurred in the AZA group and one (cervix carcinoma) in the EC-MPS group.
Discussion
There are few data on the use of non-biological agents for the management of extrarenal lupus disease. This is the first multicentre randomised long-term trial to demonstrate the superiority of EC-MPS over AZA in achieving better clinical remission rates in moderate-to-severe active non-renal lupus disease. Most patients achieved their target dose and remained in the study for the full 24 months. The study was adequately powered to assess the primary outcome, which was achieved across the treatment groups and provided valuable data of two frequently prescribed therapies in SLE.
To date, there are limited data from controlled clinical trials about the use of MMF in non-renal disease.19–24 A systematic review has identified 24 relevant studies including approximately 850 patients. Although the studies were mainly case series or open-label trials, the data suggest MMF to be effective for refractory haematological and dermatological manifestations.19–22 The only RCT by Ginzler et al 23 showed similar efficacy between MMF and cyclophosphamide in reducing non-renal disease activity, measured by BILAG score. However, results should be interpreted with caution as the study was designed for LN, high-dose corticosteroids were used as induction treatment and the BILAG index was not the primary endpoint measure in the Aspreva Lupus Management Study (ALMS).13 Our results support our hypothesis that EC-MPS would be more effective than AZA for attaining remission and maintaining clinical response during the 24-month study period. Higher rates of CR were observed as early as 12 weeks in the EC-MPS group and continued to increase over time. Secondary endpoint results including time to CR, reduction in lupus disease activity indices and the dose of corticosteroids also confirmed the superiority of EC-MPS over AZA. Although the study was not powered to demonstrate the treatment efficacy in individual organs, both study agents showed a similar profile of individual organ response except for an early remission in the cardiorespiratory domain with EC-MPS. Neurological symptoms also seemed to respond earlier in the EC-MPS group, but low sample size, the heterogeneity of neurological manifestations and the fact that some more severe patients may have been excluded prevented drawing conclusions.
We found that EC-MPS was also more effective at preventing relapses and its effect was consistent across moderate and severe flares. Moderate-to-severe SLE flares occurred in 50% of patients receiving EC-MPS compared with 71.7% of patients given AZA. Most flares were articular and mucocutaneous and in 25%–30% of cases occurred while reducing the dose of concomitant medication. Rates of severe flares were low but higher in the AZA group and mainly of haematological and renal nature. EC-MPS reduced by 45% and 65% the risk of developing any SLE flare and severe flare, respectively. The superiority of MMF over AZA in preventing LN flares has been previously reported during the maintenance phase of the ALMS.14
The occurrence of AEs and serious events was similar in both groups except for gastrointestinal side effects, including liver toxicity, and haematological events, which were more common in the AZA group, consistent with previous findings7 8 10 14 15 and were readily controlled by dose adjustments. Frequency of serious infections was low and similar in both groups. Two patients died during the study. One death occurred in each group due to pneumonia complications.
The study has some limitations. First, this is an investigator-led clinical trial with mainly Caucasian patients rather than a large international multiethnic study. Second, it was an open-label and not double-blinded trial. However, given that outcomes were strictly evaluated by an independent assessor they are unlikely to have been influenced by knowledge of patient allocation. Third, serum measures of the active metabolites of AZA (ie, 6-TGN) or EC-MPS (mycophenolic acid) were not routinely performed, leaving open the possibility that patients who failed treatment were underdosed or non-adherent to medication. Fourth, the liberty to adjust corticosteroids during the study could have confounded SLE disease activity assessments. Finally, although the trial is substantially long, potential outcomes that might appear later in time (eg, cardiovascular complications) cannot be determined.
Despite AZA being shown to be less effective, its safety profile during pregnancy is a significant advantage over EC-MPS. MMF/EC-MPS are absolutely contraindicated. MMF is likely to be a human teratogen based on the reported malformations observed among exposed offspring (microtia, orofacial clefts, external auditory canal atresia and cardiovascular malformations).36 When long-term immunosuppression is required in young women planning pregnancy this issue needs to be considered.
We conclude that EC-MPS is superior to AZA in achieving long-term clinical remission and in preventing relapse in patients with active non-renal lupus disease.
Acknowledgments
To the rest of the members of the study group: the following investigators and institutions participated in the trial: Dr Segundo Bujan Rivas, Vall d’Hebrón Hospital, Internal Medicine Department, Barcelona; Dr Sandra Parra, Internal Medicine Department, Sant Joan de Reus University Hospital, Reus; Dr Olga Capdevila i Pons, Internal Medicine Department, Bellvitge University Hospital, Barcelona; Dr José Manuel Porcel Pérez, Internal Medicine Department, Arnau de Vilanova University Hospital, Lleida; Dr Ana Belén MaDroñero Vuelta, Internal Medicine Department, Arnau de Vilanova University Hospital, Lleida; Dr Carlos Tolosa Velilla, Internal Medicine Department, Corporació Sanitària Parc Taulí, Sabadell; Dr Joaquin Oristrell, Internal Medicine Department. Corporació Sanitària Parc Taulí, Sabadell; Dr Antoni Castro Guardiola, Internal Medicine Department, Josep Trueta University Hospital, Girona; Dr Gerard Espinosa, Autoimmune Diseases Unit, Internal Medicine Department, Clinic Barcelona University Hospital, Barcelona; Dr Alberto Hernández, Internal Medicine Department, Sant Jaume Hospital, Calella; Dr Jordi del Blanco Barnusell, Internal Medicine Department, Sant Jaume Hospital, Calella; Dr Alex Vila Belmonte, Internal Medicine Department, Fundació Salut Empordà, Figueres Hospital; Dr Xavier Vidal, Clinical Pharmacology Department, Vall d’Hebrón Hospital, Barcelona; Dr Pilar Suñé, Pharmacy Department, Vall d’Hebrón Hospital, Barcelona.
References
Footnotes
Contributors JOR and JCH had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. JOR contributed to the design and execution of the study, interpretation, analysis of data and preparation of the manuscript. JCH, LSC, MPC, FM, ACS, JCP, VOS and MMP contributed to the design and execution of the study, acquisition, interpretation and analysis of data, and preparation of the manuscript. XV developed the statistical analysis plan, conducted the analysis of the data and revised the manuscript.
Competing interests None declared.
Patient consent Patient enrolled in the study signed the patient consent form approved by the Ethic Committee of Vall d’Hebron Hospital and by the Agencia Espaola del medicamento y productos sanitarios (AEMPS).
Provenance and peer review Not commissioned; externally peer reviewed.