Article Text

Abstract
Diagnosis of SLE is based on clinical manifestations and laboratory findings. Timely diagnosis and treatment are important to control disease activity and prevent organ damage. However, diagnosis is challenging because of the heterogeneity in clinical signs and symptoms, and also because the disease presents with alternating periods of flare and quiescence. As SLE is an autoimmune disease characterised by the formation of autoantibodies, diagnostic immunology laboratory tests for detecting and quantifying autoantibodies are commonly used for the diagnosis and classification of SLE. These include ANA, anti-double-stranded DNA antibodies and anti-Smith antibodies, together with other antibodies such as antiphospholipid or anti-Cq1. Complement proteins C3 and C4 are commonly measured in patients with SLE, but their serum levels do not necessarily reflect complement activation. Cell-bound complement activation products (CB-CAPs) are fragments formed upon complement activation that bind covalently to haematopoietic cells. This review focuses on the complement system and, in particular, on CB-CAPs as biomarkers for the diagnosis and monitoring of SLE, vis-à-vis complement proteins and other biomarkers of complement activation.
- Systemic lupus erythematosus
- Biomarkers
- Complement activation
- Flow cytometry
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Challenges in the diagnosis and monitoring of SLE
SLE is an autoimmune disease with alternating periods of active disease and remission that affects mainly women of childbearing age.1 Incidence and prevalence of SLE in the USA are 6 and 73 per 100 000 person-years, respectively; however, figures vary widely depending on gender, ethnicity, age and overall study methodology.2 3 SLE can biologically present with formation of autoantibodies, deposition of immune complexes in various tissues and activation of the complement system.1 This review focuses on the complement system and, in particular, on cell-bound complement activation products (CB-CAPs) as biomarkers for the diagnosis and monitoring of SLE, vis-à-vis complement proteins and other biomarkers of complement activation.
SLE diagnosis is based on clinical manifestations and laboratory findings. Clinical signs and symptoms of SLE are often non-specific and overlap with other diseases.4 This, combined with the low disease prevalence, makes the diagnosis challenging even for experienced rheumatologists. Although classification criteria set forth by the American College of Rheumatology (ACR)5 and the Systemic Lupus International Classification Clinics (SLICC)6 are not diagnostic, they can be used in clinical practice as a framework to aid in the diagnosis of SLE. Classification criteria are not widely used in the community rheumatology setting,7 and better tools are needed to aid the diagnosis of SLE, especially outside of academic centres.
Not only are classification criteria not diagnostic and not widely used to inform the diagnosis, but often patients present with signs and symptoms consistent with SLE without fulfilling the number of criteria necessary to be classified as SLE. These patients are designated as incomplete, latent or probable SLE.8 A consensus on the definition of these terms does not exist; however, the term probable SLE is used in this review to indicate patients suspected of SLE who do not fulfil the ACR classification criteria for SLE. The percentage of patients not fulfilling criteria—or patients with undifferentiated connective tissue disease—who transition to SLE over time is relatively small (approximately 10%).8 9 Various demographic, clinical and immunological features have been shown to be associated with transition to SLE, and a variable number of years may elapse before accrual of the number of criteria necessary for classification.9 10 Heterogeneity and lack of predictability add to the difficulty to diagnose SLE early, even if it is well recognised that early diagnosis and appropriate pharmacological and non-pharmacological treatment1 is critical to control symptoms of inflammation, improve quality of life, prevent organ damage due to high disease activity and, ultimately, decrease healthcare costs.11–13
The difficulties associated with SLE diagnosis suggest that biomarkers are needed to help identify and treat patients with early-stage SLE.8 9 Tests for detection and quantification of autoantibodies are commonly used for the diagnosis and classification of SLE and other autoimmune diseases. In particular, ANA, antibodies directed against double-stranded DNA (anti-dsDNA) and Smith antigen (anti-Sm), and anti-phospholipid antibodies (aPL) are important in SLE and are part of both ACR5 and SLICC6 classification criteria. ANAs are present in the vast majority of patients with SLE; however, several issues need to be considered regarding the usefulness of ANA for the diagnosis of SLE. First, ANA are also found in patients with other diseases and in healthy individuals, leading to high sensitivity but low specificity for SLE.14–16 In addition, some studies reported sensitivity of ANA in established SLE lower than expected (70%–80%),17 18 possibly because some patients may lose ANA positivity over time due to treatment or other causes. Differences in sensitivity and specificity may also be related to the assay employed, as different ANA assays can give different results.17 As ANA positivity is part of the SLE classification criteria and is commonly performed to aid the diagnosis of SLE, ANA test results need to be interpreted with caution. In contrast, anti-dsDNA and anti-Sm antibodies have high specificity but low sensitivity for SLE.14 15 Similar to ANA, assays for anti-dsDNA antibodies lack standardisation.19 20 aPL are a heterogeneous subset of autoantibodies directed against protein–phospholipid complexes. They are common in SLE; however, sensitivity is low6 and assays have limitations due to lack of standardisation and poor reproducibility.21
Numerous instruments have been developed and validated to assess SLE disease activity and organ damage. The most commonly used instruments are SLE Disease Activity Index (SLEDAI), of which several versions have been developed; the Systemic Lupus Activity Measure (SLAM); the European Consensus Lupus Activity Measure (ECLAM); the British Isle Lupus Assessment Group Index (BILAG); and the SLICC/ACR Damage Index (SDI) for SLE.22 In addition to these instruments, patient-reported outcome measures (PROMs) could offer a pragmatic quantitative measure of patient well-being over time. The patient perspective is increasingly recognised to be important, and numerous PROMs, some specific for lupus and some generic, have been developed and are being used in clinical studies.22 Although the importance of monitoring disease activity over time is also well recognised in clinical practice, disease activity instruments have limitations, including no change in the overall score when some manifestations improve and others worsen, and that a similar score in different patients may result from involvement of different organs.22 In addition, the use of SLE indices requires training to complete and interpret accurately, and these instruments are not routinely used in most clinical settings. ANA testing is not useful for monitoring SLE disease activity16 and, in fact, is not part of disease activity indices. Conversely, anti-dsDNA antibodies have been shown in some studies to correlate with disease activity and kidney involvement; however, conflicting results have been reported.20 Also complement protein levels are included in disease activity instruments; however, the limitations discussed in the next section render these markers suboptimal. It follows that better biomarkers are needed as an objective means to monitor disease activity.23 24
The complement system in SLE
The involvement of the classical complement system in the pathogenesis of SLE is well established. While complement deficiencies predispose to SLE, activation of the classical complement system by autoantibodies and immune complexes contribute to inflammation and tissue injury.25
The complement system consists of more than 30 soluble blood proteins that are part of the innate immune system. C3 is the most abundant complement protein, with normal plasma concentration in humans of 90–180 mg/dL; C4 is the second abundant, with normal concentrations of approximately 12–50 mg/dL.
Complement activation occurs through three pathways: classical, alternative and lectin pathways.25 26 The classical pathway is activated when C1q in the C1 complex (formed by C1q, C1r and C1s) binds to the Fc portion of antibodies or antigen–antibody complexes. On binding of C1q, C1r and C1s are activated, and C1s cleaves C4 and C2 to form the C3 convertase, C4b2a (figure 1). The alternative pathway is initiated when spontaneously hydrolysed C3 (called iC3 or C3(H2O)) is in the proximity of cell surface molecules of invading bacteria or other pathogens (lipopolysaccharide on gram-negative bacteria, teichoic acid on cell walls of gram-positive bacteria, zymosan on fungal and yeast cell walls) or self-tissue. The lectin pathway is similar to the classical pathway, but instead of being triggered by antibodies, the lectin pathway is initiated when mannose-binding lectins (MBL) attach to carbohydrates on the surface of microorganisms. MBL-associated serine protease (MASP) is activated and cleaves C4, leading to the formation of C4a and C4b; MASP also cleaves C2, ultimately leading to the formation of the C3 convertase C4b2a.
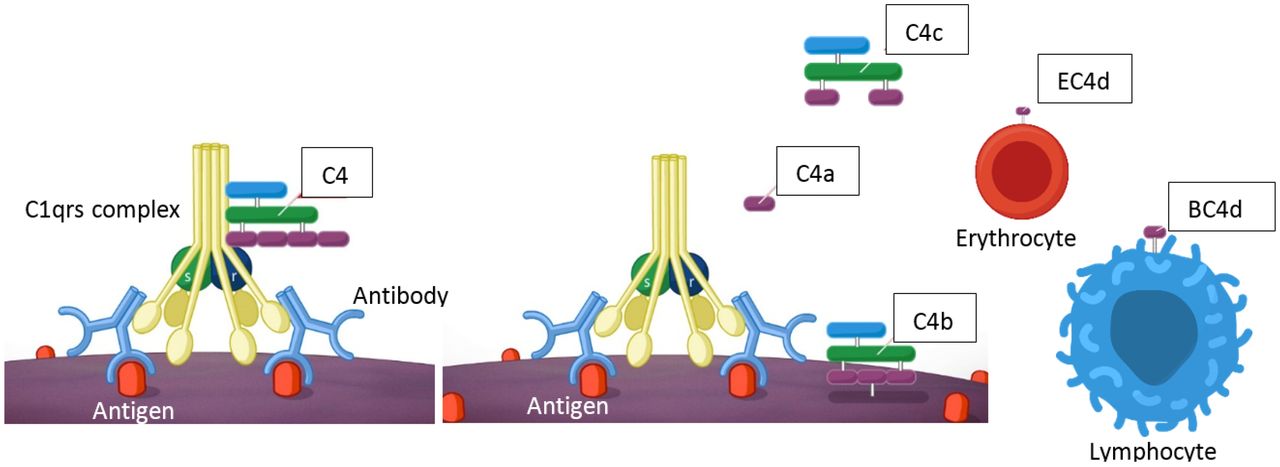
The classical complement cascade is activated on binding of the C1 complex to the Fc stem of antibodies bound to their antigens. C1s in the C1 complex activates C4, which is cleaved into the anaphylatoxin C4a and the C4b fragment. C4b binds to cell surfaces and is further processed into C4c and C4d, which remains bound to cells. The right-hand side of the figure shows C4d bound to erythrocytes (EC4d) and lymphocytes (BC4d or TC4d). C3 is activated downstream of C4 by the C3 convertases. Although not shown in the figure, C3 activation also leads to the formation of the soluble anaphylatoxin C3a while C3b binds to cells. C3b is further processed and C3d remains bound to cell surfaces.25 26 29 See the text for additional details.
The three pathways culminate with the activation of C3 and the formation of the C5 convertases (C3b2Bb in the alternative pathway and C4b2a3b in the classical and lectin pathways), which convert C5 into C5a and C5b. C5b recruits C6, C7, C8 and C9 leading to the assembly of the membrane attack complex (MAC). The MAC causes cell lysis and death26 27 or cellular activation at sublytic concentrations.28
C3a, C4a and C5a are soluble polypeptides called anaphylatoxins. They bind to complement receptors and promote inflammation and tissue damage by numerous mechanisms, including attracting phagocytes (neutrophils and macrophages) and promoting vasodilation and release of histamine from mast cells.26
C3 and C4 are formed by two and three polypeptide chains, respectively, linked by disulfide bonds.29 C4 is activated by C1s and is converted into C4a and C4b, while C3 is activated by C3 convertases (C4b2a in the classical and lectin pathways, and C3bBb in the alternative pathway) and is converted into C3a and C3b. Activation of C4 and C3—with formation of C4b and C3b—is brought about by the cleavage of a peptide bond; the resulting conformational change exposes a thioester bond that can be hydrolysed or can react with nearby macromolecules. In the latter case, C4b and C3b bind covalently to cellular components in the immediate surrounding, such as hydroxyl groups of carbohydrates and amino groups of proteins of pathogens, host cells or immunocomplexes.29 Once formed, C4b and C3b are further degraded by the serine protease factor I and cofactors such as membrane cofactor protein (MCP) (CD46), complement receptor 1 (CR1) (CD35) and factor H.26 C4b is cleaved into C4c and C4d, with C4d remaining bound to cell surfaces (figure 1). C3b is processed into iC3b and C3f; iC3b is cleaved into C3c and C3dg; finally, C3dg is cleaved into C3d and C3g, with C3d remaining bound to cell surfaces. C4d is approximately 45 kDa and C3d is approximately 35 kDa.29
Genetic deficiency of certain complement proteins (in particular C1q, C1r/C1s, C4, C3, C2, etc) or a low copy number of the two genes for C4, C4A and C4B, has been shown to predispose to the development of SLE.24 27 30 Several hypotheses have been formulated to explain this link. According to the so-called waste disposal hypothesis, complement may help clear immune complexes and apoptotic cells, which are considered the main source of autoantigens in SLE. Alternatively, the complement system may promote tolerance by facilitating the negative selection of self-reacting lymphocytes.31 A third hypothesis is that complement may regulate synthesis of cytokines involved in SLE pathogenesis, such as type I interferons.27 30
While complement deficiencies may participate in the pathogenesis of SLE, complement dysregulations may be present also during the course of the disease. This has led to the use of complement as a biomarker of SLE. As the classical complement system is activated by autoantibodies and immune complexes during active disease, C3 and C4 may be consumed. Indeed, patients with SLE may present with low C3, low C4, or decreased haemolytic activity of the complement system (low CH50). These lab abnormalities, although absent in the ACR classification criteria,5 have been included in the SLICC criteria because of their clinical importance.6 Other complement proteins are not routinely measured as biomarkers of SLE because of their relatively low plasma concentration. However, measurement of C3 and C4 as SLE biomarkers has several drawbacks. In particular, the range of C3 and C4 in normal plasma is wide and overlaps with the range observed in many patients with SLE. Because of the high concentrations of these proteins (especially C3), a small change in their levels may be difficult to detect and may not lead to values below the normal range. In addition, consumption of C3 and C4 during complement activation can lead to increased synthesis to counteract the increased consumption, leading to no net change in protein levels. Overall, the levels of total C3 and C4 do not necessarily reflect complement activation, and an increase in soluble complement split products, such as C3a and C4a, may be more physiologically relevant to indicate complement activation. However, stability issues make the measurement of soluble split products challenging in normal clinical practice, and they have not replaced C3 and C4 as biomarkers of SLE.28
The limitations associated with the measurement of complement proteins and soluble split products have prompted the study of CB-CAPs as biomarkers of complement activation in SLE.
CB-CAPs in SLE diagnosis
CB-CAPs are complement split products bound to blood cells and include C4d bound to B and T cells (BC4d and TC4d, respectively), erythrocytes (EC4d), reticulocytes (RC4d) and platelets (PC4d). CB-CAPs can be quantified by flow cytometry. Extensive research by several groups has demonstrated that cell-bound C4d and C3d are elevated in patients with SLE compared with other diseases and may have value for the diagnosis and monitoring of patients with SLE.28 32–35
Initial work focused on EC4d because of the abundant number of erythrocytes in blood. EC4d levels, measured by flow cytometry, were significantly higher in patients with SLE than in those with other diseases or healthy volunteers.32 Assessment of other CB-CAPs in patients with SLE and other diseases overall showed that CB-CAPs are sensitive and specific markers of SLE, and that CB-CAPs can be used to aid the diagnosis and monitoring of SLE.28 PC4d is the CB-CAP with the highest specificity (up to 100% in some studies), although sensitivity is relatively low. Sensitivities and specificities of CB-CAPs are reported in table 1. The different values reported in different publications can be attributed to how the cut-off values were established or to the different patient populations studied.
Sensitivity and specificity of EC4d, BC4d, TC4 and PC4d
Interestingly, if a patient is positive for a certain CB-CAP (eg, PC4d or TC4d), all cells of that particular cell type are coated with C4d, indicating that all circulating cells, including the newly synthesised, are C4d positive. In addition, C4d is deposited on cells in a homogeneous manner, suggesting that C4d generated during complement activation binds to multiple moieties of the cell surface or, alternatively, that the cell membrane undergoes changes that lead to a homogeneous deposition of C4d.33 36 Because erythrocytes circulate for approximately 90 days, different subpopulations of erythrocytes may express different levels of EC4d; erythrocytes circulating during a flare may be coated with more C4d molecules than erythrocytes synthesised at a later time point when the flare has subsided.28
Complement activation and formation of CB-CAPs does not necessarily lead to the formation of the MAC and cell lysis. In fact, TC4d-positive patients do not present with lymphopoenia.36 However, CB-CAPs deposition may lead to deformation of the cell membrane and/or cell activation.37 38
A study on PC4d showed that PC4d-positive patients were negative for antiplatelet IgG and IgM, suggesting that antiplatelet antibodies are not responsible for complement activation and C4d deposition on platelets.33 On the other hand, an association between positivity for PC4d and aPL was found, suggesting that aPL may be in part responsible for complement activation and formation of PC4d.33 aPLs amplify platelet activation and activated platelets, in turn, activate the complement system leading to deposition of complement split products on their surface. Platelets in patients with SLE can be activated in the absence of aPL, and both platelet activation and aPL can independently mediate complement activation on platelets and deposition of CB-CAPs.35 39 To what extent these phenomena contribute to the increased risk of arterial or venous thrombosis in SLE is not completely understood (figure 2).
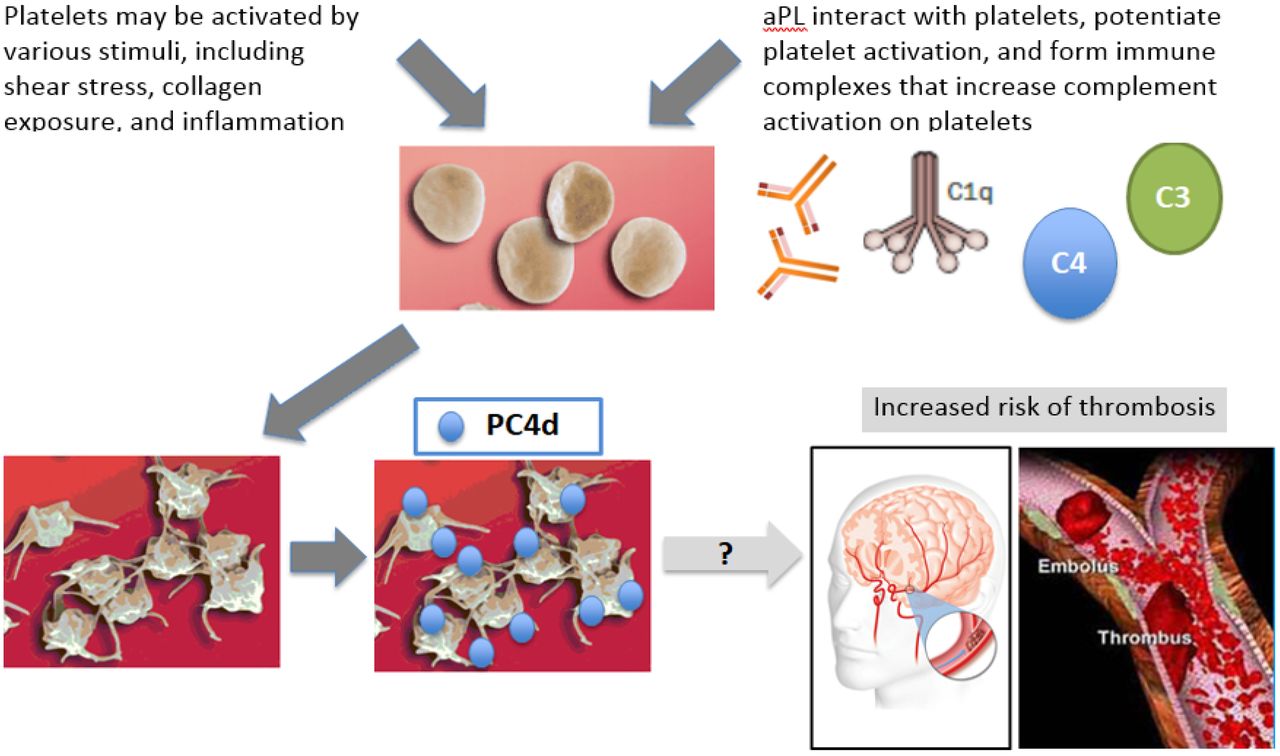
Platelet can be activated in SLE by various stimuli, including shear stress, collagen exposure and inflammation. aPL may contribute to platelet activation. Formation of immune complexes may lead to complement activation and deposition of C4d on the platelet surface. How these phenomena contribute to the increased risk of cardiovascular disease in SLE is not completely understood.35 39 50–52 See the text for additional details. aPL, antiphospholipid.
In contrast to the absence of antiplatelet antibodies in PC4d-positive patients, anti T-cell antibodies were found on T cells of approximately 30% of TC4d-positive subjects with SLE.36 The antigens of these antibodies have not been clearly identified.36 Taken together, these data lead to the hypothesis that the dynamic binding of various autoantibodies to cells causes complement activation and deposition of CB-CAPs on those cells. This is consistent with the observation that not all cell types are simultaneously C4d or C3d positive in a given patient, and that CB-CAPs bind to different cell types in a patient-specific fashion.36
C3d and C4d bind covalently to cell surfaces and remain attached for the lifespan of the cell. This stability allows to reliably measure EC4d and BC4d in a clinical laboratory for up to 2 days after phlebotomy, differently from soluble complement split products.40 The favourable pre-analytical characteristics make EC4d and BC4d attractive as candidate biomarkers of SLE. When thorough quality control systems are implemented,40 these markers can be measured reliably in a clinical laboratory at a remote location.
In addition to this practical aspect, EC4d and BC4d have better diagnostic characteristics than complement proteins C3 and C4 or anti-dsDNA, as demonstrated in a large multicentre study (table 2).14 In fact, the combination of reduced C3 and C4 was 45% sensitive for SLE with specificity ranging from 88% to 96% versus other diseases. As expected, anti-dsDNA had low sensitivity and high specificity (sensitivity of 33% and specificity between 93% and 100% vs other diseases). Elevated CB-CAPs (EC4d>14 net mean fluorescence intensity (MFI) and/or BC4d>60 net MFI, both cut-offs established based on the 99th percentile of normal healthy volunteers) was 66% sensitive with overall specificity of 85% versus other diseases and of 99% versus healthy volunteers.14
Sensitivity and specificity of EC4d and BC4d in SLE have been further improved by the addition of other markers to yield a panel with adequate performance for its use in clinical practice. A multi-analyte assay with algorithm was developed that combines EC4d and BC4d with ANA, anti-dsDNA, anti-Sm and autoantibodies associated with diseases other than SLE (in particular, anti-citrullinated peptide autoantibodies, anti-SS-B/La, anti-CENP, anti-Jo-1 and anti-Scl-70 antibodies) that together form the so-called specificity component. The panel’s two-tier assessment, which has been described in detail elsewhere,7 14 40 demonstrated high sensitivity and specificity in differentiating patients with SLE from those with other diseases. In particular, the two-tier model achieved 80% sensitivity for SLE with a specificity of 86% versus other diseases,14 98% versus healthy volunteers14 and 100% versus patients with fibromyalgia.18 In addition, the two-tier model demonstrated good performance characteristics, as compared with the physician’s diagnosis, in a retrospective chart review study conducted by community rheumatologists.7
It is well established that CB-CAPs have higher sensitivity than C3/C4 in patients fulfilling classification criteria, and limited data suggest that they may also help aid the diagnosis of patients with probable SLE. For example, a recent case report described a difficult to diagnose patient with thrombocytopaenia, myelitis, ANA and lupus anticoagulant. The patient was negative for anti-dsDNA, anti-Sm, anti-RNP or anti-cardiolipin antibodies; in addition, C3 and C4 were in the normal range even at the time of myelitis. Interestingly, the patient was positive for anti-aquaporin-4 antibodies, the autoantibodies found in patients with neuromyelitis optica. The positivity for EC4d and BC4d supported the diagnosis of SLE in this patient, and informed therapeutic decisions.41 Additional studies are required to establish the performance characteristics of CB-CAPs in patients with probable SLE or in patients otherwise suspected of SLE who do not fulfil current classification criteria.
CB-CAPs in SLE disease activity and disease monitoring
Assessment of disease activity is challenging in SLE because of the complexity and variability in clinical presentation over time. However, treatment and close monitoring of patients with SLE is important to induce or maintain remission and prevent organ damage. Biomarkers could facilitate monitoring of patients with SLE. Anti-dsDNA antibodies have been studied extensively; however, their ability to predict renal flares is uncertain.42 Antibodies against the first protein of the classical complement pathway, C1q, are associated with lupus nephritis, and several other biomarkers have been studied as markers of disease activity or organ involvement.23 Because of the importance of complement activation in SLE, sequential measurements of C3 and C4 can be used to monitor disease activity; however, the drawbacks discussed earlier may limit the usefulness of these markers, at least when used in isolation.
CB-CAPs have been studied as markers of SLE disease activity. C4d bound to reticulocytes can provide a snapshot of disease activity: as reticulocytes are short lived, the presence of C4d on their surface may reflect current disease status.43 44 In addition, correlation between CB-CAPs and disease activity indices has been recently investigated.14 34 45
High CB-CAPs (EC4d and/or BC4d) and the two-tier model discussed above had higher sensitivity, compared with anti-dsDNA antibodies and low C3/C4, in particular in patients with SLE with low disease activity measured with the commonly used index SELENA-SLEDAI. This suggests that CB-CAPs may be particularly important in aiding the diagnosis of patients with early or mild SLE (figure 3).14

{kind=link}
{kind=link}
{kind=link}
Positivity rate for anti-dsDNA antibodies, low complement proteins C3 and/or C4, high CB-CAPs (EC4d > 14 net MFI and/or BC4d > 60 net MFI) and two-tiered methodology stratified by Disease Activity Score determined using the nonserological (without anti-dsDNA and low complement) SELENA-SLEDAI score. The number of patients in each of the nonserological SELENA-SLEDAI category is indicated in the X axis. Reproduced from Putterman et al.14 http://creativecommons. org/licenses/by nc/4.0. CB-CAP, cell-bound complement activation product; ds-DNA, double-stranded DNA; SELENA-SLEDAI, Safety of Estrogens in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index SELENA Modification.
EC4d and EC3d have been investigated as markers of disease activity measured with the SELENA-SLEDAI and the SLAM.34 Not only EC4d and EC3d were higher in patients with SLE than in patients with other disease and healthy volunteers, but these CB-CAPs were higher in patients with SLE with higher disease activity at the time of blood draw. In addition, EC3d and EC4d correlated significantly, although weakly, with C3 and C4, indicating that EC3d, EC4d, C3 and C4 could provide additive information to evaluate disease activity.34
Consistent with these results, a prospective study that enrolled patients with active disease and elevated CB-CAPs showed that EC3d, EC4d, anti-dsDNA and anti-C1q antibodies decreased with improvement of disease activity, while C3 and C4 increased. Increases in C3 and C4 and decreases in EC4d and anti-C1q were also associated with reduction of proteinuria, while changes in anti-dsDNA antibodies and EC3d were not.45Patient-reported outcomes were also evaluated using the Short Form-36 (SF-36) questionnaire at every visit. Reduction in EC4d and EC3d was associated with improvement of six of the eight domains of the SF-36, while other markers were associated with two or fewer domains, indicating that changes in CB-CAPs may be better correlated with patient well-being than more traditional biomarkers, for example, complement proteins and anti-dsDNA antibodies.45
A small study that evaluated patients with lupus nephritis, patients with SLE without renal involvement and patients with renal disease due to conditions other than SLE showed that levels of EC4d and RC4d were higher in the lupus nephritis group than in the other two groups, suggesting a potential role of cell-bound C4d as a marker for lupus nephritis.46 The same study reported that SLE patients with or without nephritis were positive for PC4d (43% and 30%, respectively), while no patient with kidney disease not due to SLE such as diabetes was PC4d positive,46 confirming the high specificity of PC4d in SLE observed in other studies.33
A pilot study that enrolled patients with low disease activity and followed them prospectively showed that EC4d levels were higher at the visits when disease activity was higher,47 providing additional evidence that EC4d and/or other CB-CAPs, possibly in combination with other biomarkers, could be useful to monitor disease activity in patients with SLE.
Conclusions
SLE is a heterogeneous disease with alternating periods of quiescence and exacerbation. If not appropriately managed, SLE can lead to significant organ damage. However, diagnosis and monitoring of disease are often challenging. CB-CAPs, other biomarkers and assay panels may aid the diagnosis and monitoring of patients with this disease.
References
Footnotes
Competing interests Dr R-G is a site investigator for Exagen Diagnostics. Drs RVA, TD are employees of Exagen Diagnostics. Mr. JL has no competing interests.
Provenance and peer review Commissioned; internally peer reviewed.
Data sharing statement Data sharing is not applicable for a review article.