Article Text

Abstract
Objective SLE serves as an independent risk factor` for endothelial dysfunction (ED) not explained by Framingham risk factors. We sought to understand the development of SLE-induced ED on a cellular level in order to develop strategies aimed at reversing cellular abnormalities. This study assessed the impact of SLE patient serum on endothelial nitric oxide synthase (eNOS), nitric oxide (NO) production and functional changes in the cell.
Methods Human umbilical vein endothelial cells (HUVECs) cultured in serum of either SLE (n=25) or healthy patients (n=14) or endothelial basal medium 2 (EBM-2) culture media supplemented with fetal bovine serum with or without L-sepiapterin were used for our studies. We applied the fluorescent probe DAF-FM diacetate for intracellular NO detection using flow cytometry. Total RNA isolates were analysed using reverse transcription PCR for eNOS mRNA expression. Oxygen consumption rate was determined using seahorse analysis. Neutrophil adhesion and migration were determined using a calcein AM microscopy assay.
Results The mRNA expression of eNOS was increased in SLE cultured HUVECs compared with healthy control (p<0.05). The SLE eNOS mRNA level correlated with SLE patient age (p=0.008); however, this trend was not observed with healthy patients. SLE serum reduced NO production in HUVECs compared with EBM-2 cultured cells (p<0.05). Co-treatment of endothelial cells with L-sepiapterin preserved HUVEC capacity to produce NO in SLE conditions (p<0.01). SLE serum enhanced neutrophil migration (p<0.01) but not neutrophil adhesion compared with healthy controls. The bioenergetic health index was not different.
Conclusions SLE likely causes disruption of endothelial cell eNOS function and NO modulated pathways.
- nitric oxide
- systemic lupus erythematosus
- endothelial dysfunction
- endothelial nitric oxide synthase
- L-sepiapterin
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- nitric oxide
- systemic lupus erythematosus
- endothelial dysfunction
- endothelial nitric oxide synthase
- L-sepiapterin
Introduction
SLE is a heterogeneous chronic autoimmune inflammatory syndrome that predominately impacts women of childbearing age. Although specific clinical and immunological criteria have been defined, its clinical course is highly variable with some patients experiencing life-threatening cardiovascular disease complications which account for one-third of deaths in patients with SLE.1 2 A significant proportion of patients with SLE display accelerated endothelial dysfunction which precedes cardiovascular disease (CVD). The endothelium plays a pivotal role in governing vascular function and, thus, prevents the development of vascular abnormalities.3–5 Chronic inflammation promotes endothelial cell injury leading to generation of superoxide and expression of cell adhesion markers.6 7 These perturbations in endothelial cell function promote oedema, leucocyte trafficking and organ damage. While causes of endothelial dysfunction in SLE are multifactorial, the specific biological underpinnings governing the development of endothelial dysfunction in SLE are incompletely understood and represent an important area of research.
Nitric oxide (NO) is a membrane-permeable free radical, continuously synthesised by the endothelial nitric oxide synthase (eNOS) dimeric enzyme.8 Diffusion of NO across the cellular membrane as a paracrine mediator has impacts on cellular function critical for blood vessel dilation and unobstructed blood flow.9–11 Diminished release of NO and loss of eNOS expression have been consistently linked to endothelial dysfunction.12 13 Paradoxically, uncoupled eNOS, likely resulting from oxidation of tetrahydrobiopterin (BH4), leads to eNOS enzymatic dysfunction.14–16 Biopsies from patients with severe lupus nephritis show diminished eNOS expression.17 18 Accordingly, our previous work demonstrated that genetic ablation of eNOS in lupus-prone MRL/lpr mice resulted in accelerated, more severe disease with significant declines in survival.19 MRL/lpr mice lacking eNOS display increased superoxide production associated with increased MCP1 production, increased glomerular crescentic and necrotic lesions, and reduced levels of the anti-inflammatory cytokine interleukin (IL)-10.20 Accordingly, a positive correlation between H2O2 and eNOS levels in vitro exists.21 NO produced in the presence of O2.- yields peroxynitrite (ONOO-) which oxidises the essential eNOS co-factor BH4 to dihydrobiopterin (BH2) and biopterin. This oxidation of BH4 leads to uncoupling of the eNOS homodimer to monomers, resulting in reactive oxygen species (ROS) rather than NO production.22 23 ROS production itself leads to endothelial dysfunction. L-sepiapterin (L-sep) is a precursor for tetrahydrobiopterin (BH4) synthesis, and previous studies have shown its efficacy in restoring eNOS function, possibly through a recoupling mechanism.24 Thus, L-sep may serve as a viable therapeutic option when eNOS uncoupling is the predominant mechanism of endothelial dysfunction in an inflammatory microenvironment, such is present in patients with SLE.
A hallmark of endothelial inflammatory responses which coincide with diminished endothelial-NO is increased neutrophil migration and adhesion to the endothelial cell surface.25 26 Neutrophil migration and adhesion are processes that promote progression of atherosclerosis through the release of myeloperoxidase, promoting ROS production, and sequestering monocytes to the intravascular space.27 Recent studies link NETosis, a process of neutrophil death emerging as a potential pathogenic process in SLE, to atherosclerosis.28 In the present study, we used human umbilical vein endothelial cells (HUVECs) to study the impact of SLE serum on eNOS expression and NO production in vitro. We hypothesised that SLE serum would negatively impact NO production in HUVECs, leading to enhanced leucocyte adhesion and migration.
Patients and methods
Patient population
Specimens for this study were stored and collected from study visits that were part of a longitudinal observational cohort study known as the SLE Gullah Health or SLEIGH study initiated in 2002 by Dr. Diane Kamen and has been previously described.29 All patients classified as having SLE met 4 of the 11 classification criteria as specified by the 1997 American College of Rheumatology criteria.30
Clinical characteristics of the study population
Patients with SLE were evaluated during regular clinical visits by clinicians trained in SLE disease activity measures. The clinical and laboratory elements of the SLE Disease Activity Index (SLEDAI) were recorded if they were attributed to SLE disease activity. The scores were recorded for each blood collection visit. Control volunteers were evaluated for autoimmune disease and the presence of cardiovascular disease or risk factors (hypertension, smoking, hypercholesterolemia or previous myocardial infarction, cardiac or brain revascularisation, or stroke).
Blood collection
Blood from healthy and participants with SLE were collected in a sterile vacutainer blood collection tube and whole blood was allowed to clot at 25°C for 10 min. Samples were centrifuged to remove the clot and remaining serum was stored in aliquots at −80°C for future use.
Neutrophil isolation
Neutrophils were isolated as previously described.31 Briefly, 20 mL of human blood was acquired from healthy volunteers and cells were isolated using Lymphocyte Separation Medium (Cellgro, Manassas, Virginia, USA). The assay was validated based on forward scatter and side scatter using flow cytometry (data not shown).
Endothelial cell culture and serum culturing
Primary human umbilical vein endothelial cells from pooled donors were purchased from Lonza (Walkersville, Maryland, USA) and cultured according to the manufacturer’s instructions in 5% CO2 at 37°C in humidified air. Cells were cultured in endothelial cell basal medium-2 (EBM-2) supplemented with EBM-2 SingleQuot (Lonza), pH 7.6–8.0. Media was changed every other day until cells were 70%–80% confluent. HUVECs were subcultured using TryPLE Express, pH 8.0 (ThermoFisher Scientific, Waltham, Massachusetts, USA). Cell growth was limited to 12 population doublings, and all experiments were carried out using cells between passages 3–5. For serum experiments, cells were cultured with 20%–50% serum for 6 or 24 hours prior to further analysis.
Real-time reverse transcription-PCR (RT2PCR)
To detect changes in mRNA levels, HUVECs were treated with 20% SLE or healthy sera for 6 or 24 hours as specified. Cells were harvested following treatment and total RNA was extracted using a Trizol (ThermoFisher Scientific)-RNeasy kit (Qiagen, Frederick, Maryland, USA) hybrid protocol as previously described.32 RNA integrity was assessed using a NanoDrop 2000c UV-Vis spectrophotometer (ThermoScientific, Wilmington, Delaware, USA) and samples with A260/280 ratios of 1.8–2.1 were used. Single strand cDNA was synthesised from 1 µg of RNA using an iScript cDNA synthesis kit (Bio-Rad, Hercules, California, USA). For each reaction, 1 µL of cDNA product was used for signal amplification with SsoAdvanced universal SYBR (BioRad). A CFX96 Real Time PCR Detection System (Bio-Rad) was used to assess changes in NOS3 and GAPDH, using commercially available primers (Qiagen). The relative expression was calculated using the equation 2-ΔΔCt (Δ; experimental gene cycle threshold (Ct) – housekeeping gene (Ct)). The fold change gene expression of interest was calculated based on normalisation to GAPDH . PCR was performed ≥3 independent experiments with at least three replicates.
Measurement of nitric oxide production
For real-time detection of NO production in HUVECs, 1.2×105 cells were seeded in a 12-well tissue culture plate. Following adherence, cells were serum starved for 6 hours in endothelial basal media (EBM) containing 0.2% fetal bovine serum (FBS). Cells were stimulated with either 50% healthy or SLE sera ± L-sep (5 µM; 6 hours), the eNOS-specific inhibitor, Nω-Nitro-L-arginine (L-NNA, 10 µM; 30 min pre-incubation (Tocris; Bristol, UK)) or the NO donor 3,3ʹ-diamino-4ʹ-methoxyflavone (DD1, 10 µM, Tocris). Following stimulation, cells were washed twice with phosphate buffered saline (PBS) and loaded with 1 µM DAF-FM diacetate (4-amino-5-methylamino-2’,7’-difluorofluorescein diacetate, 1 µM) (ThermoFisher Scientific) in phenol red-free EBM for 30–45 min. Cells were washed twice with PBS and dissociated from plates using phenol-red free TryPLE Express (ThermoFisher Scientific) and fixed with 2% paraformaldehyde for 3 min. A population of 2000–10 000 cells were gated to remove doublets and controls and analysed based on their fluorescence intensities using a FACS Calibur flow cytometer (Becton Dickenson, San Diego, USA). The mean fluorescence intensity (MFI) was normalised to respective populations in unstimulated cells. In order to discriminate between NO and other gaseous molecules previously shown to augment DAF-FM fluorescence, we performed a urate assay to optimise our assay (data not shown).
Oxygen consumption
Endothelial cells were seeded at 20 000 cells/well on a Seahorse 96-well XF Cell Culture Microplate as detailed by the manufacturer (Seashore Bioscience/Agilent Technologies, Santa Clara, California, USA) and allowed to adhere overnight in complete EBM-2 (EBM-2 basal media plus EBM-2 SingleQuots, Lonza, Basel, Switzerland). The following day cells were rinsed with 1× PBS and 50% control or SLE patient serum was added to wells and allowed to incubate for 24 hours (six samples per group with five replicates per patient sample). The Seahorse XF Analyzer (Seashore Bioscience/Agilent Technologies) was used to determine basal oxygen consumption rate (OCR). Four basal rate measurements were followed by four measurement cycles following each injection (1 µM oligomycin, 1 µM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone and 2 µM AA rotenone). Consumption rates were calculated as previously described.33 The bioenergetic health index (BHI) was calculated using the following formula: BHI= (ATP-linked × Reserve Capcity)/(Proton Leak × Non-mitochondrial OCR).34
Neutrophil adhesion assay
HUVECs were plated at 5.0×104 cells/mL in a 24-well plate (Costar) and allowed to adhere overnight. HUVECs were serum starved for 3 hours in phenol-red free 0.2% FBS EBM media (Lonza) prior to activation with 10% sera for 4 hours. Tumour necrosis factor-α (100 ng/mL) was used as the positive control. Neutrophils isolated from healthy human blood as outlined previously were labelled with Calcein AM (Life Technologies) at 5×105 cells/mL. Neutrophils were washed gently four times in warm serum-free EBM culture media prior to co-culturing with HUVECs for 60 min after which non-adherent cells were removed by repeated gentle washing (four times) with EBM culture media. Fluorescence intensity was measured at 520 nM with a FLUOStar Omega microplate reader (Cary, North Carolina, USA), and images were captured using confocal microscopy. Data are reported as ratios of the number of neutrophils to the number of endothelial cells as averages from three different visual fields.
Neutrophil migration assay
Transwell migration assays were performed as described elsewhere.35 Briefly, transwell inserts (3 µm pore) were pre-coated with fibrinogen and allowed to incubate overnight for 24 hours. HUVECs were seeded in 24-well plates at 1.0×104 cells per well and allowed to adhere overnight. Cells were activated with 50% sera from healthy and SLE controls for 6 hours and washed once in PBS. Neutrophils were added to chambers and inserted into media ± IL-8 (1.25 nM, Cell Signaling). After 60 min, the number of neutrophils in the lower chambers was visualised using 4× magnification and quantified using confocal microscopy. All values were normalised to untreated controls.
Statistical analysis and data handling
Descriptive statistics are reported as mean±SD or IQR for continuous variables. Gaussian distribution was determined using the D’Agostino-Pearson omnibus normality test and the Shapiro-Wilk normality test. Paired and unpaired (where appropriate) two-tailed Student’s t-test and non-parametric Mann-Whitney test were used on non-parametric data analysis on lupus and controls. Correlations were determined using Pearson’s or Spearman’s correlation analysis and are reported accordingly. Standardised univariate regression analysis was performed to adjust for lupus-associated indicators of disease activity and β-coefficients and p values were reported. Analysis of variance test with Tukey’ Fisher’s probable least significance post-test was used to analyse NO and neutrophil adhesion data. No mathematical correction was made for multiple comparisons. Data are presented as mean±SEM. Differences were considered significant if the p value was ≤0.05. Statistical analysis was performed using IBM SPSS Software V.25 or GraphPad Prism V.6.0f (San Diego, California, USA).
Results
Demographic and clinical characteristics of study participants
SLE (n=25) and healthy controls (n=14) did not differ in sex, race or age. Mean±SD disease duration for patients with SLE was 9.4±6.4 and the age at diagnosis was 22.6±9.1. SLEDAI36 37 was 4.6±3.5, reflecting overall mild lupus disease activity. Complement C3 levels were 104.7±37.4 while C4 levels were 24.3±13.8 further supporting the notion that patients with lupus in this study had mild disease activity. Patients with SLE had normal blood pressures and an overall body mass index of 26±5, indicating a group of participants that were slightly overweight. Most patients with SLE were taking prednisone and/or antimalarial medications (76%, 68%, respectively) at the time of the study visit (tables 1 and 2).
Demographics
Demographics and clinical characteristics
Serum from patients with SLE alters eNOS mRNA expression
We determined whether eNOS mRNA expression changed in primary HUVECs in response to 50% (v/v) serum of EBM-2, control patients or patients with SLE. SLE serum increased eNOS mRNA expression compared with healthy patient serum (1.88±0.22-fold, p<0.05; figure 1A). SLE patient age but not control patient age negatively correlated with eNOS mRNA expression in SLE serum cultured HUVECs (r=−0.59, p=-0.003; figure 1B). The correlation persisted in multivariate regression analysis adjusting for lupus disease activity as measured by SLEDAI (figure 1C), disease duration, dsDNA, and C3 and C4 levels (data not shown). None of these disease activity covariables themselves independently associated with eNOS mRNA expression (data not shown). No significant correlation was found between eNOS mRNA and SLE patient blood pressures (figure 1D). No other correlations with eNOS mRNA expression were observed.
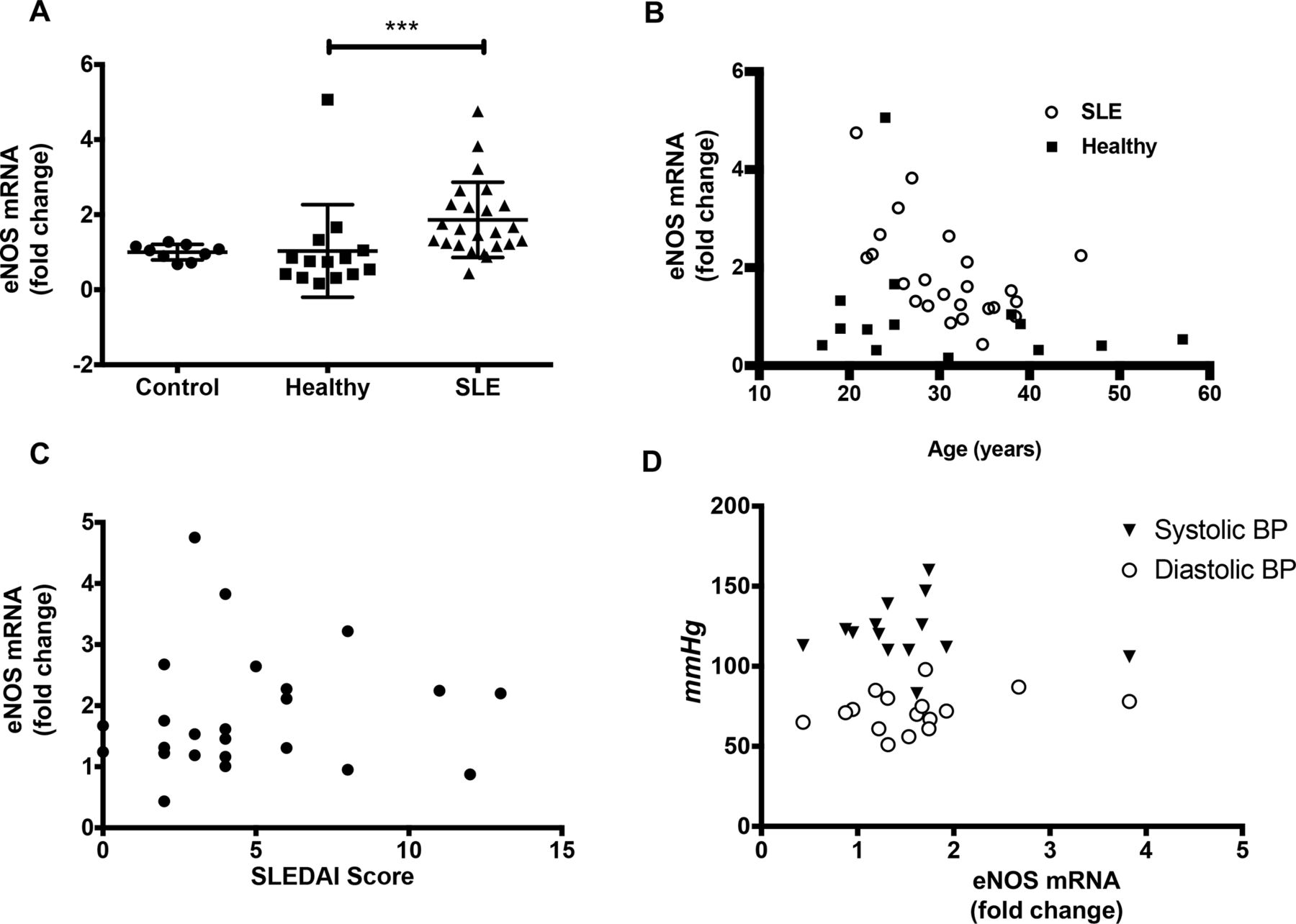
SLE sera-induced endothelial nitric oxide synthase (eNOS) mRNA expression in human umbilical vein endothelial cells. (A) eNOS mRNA levels from human umbilical vein endothelial cells (HUVECs) treated with buffer 20% control or SLE sera were as follows: for buffer controls (9), 1±0.06918; for healthy controls (14),1.032±0.3294; for SLE (22) 1.888±0.2229 (mean±SEM). (B) Lupus-induced eNOS mRNA levels correlate with patient age but not control derived eNOS mRNA, p<0.01. (C) Lack of association between eNOS mRNA and SLE Disease Activity Index (SLEDAI) scores. (D) Lack of association between NOS3 expression and systolic blood pressure (BP) or diastolic BP measured in millimetre of mercury. *p<0.05, **p<0.001, ***p<0.0001. Samples were analysed using a Kruskal-Wallis non-parametric multiple comparisons test and a Dunn’s post-test and Spearman’s correlation. r=rho value.
SLE serum reduced NO in endothelial cells
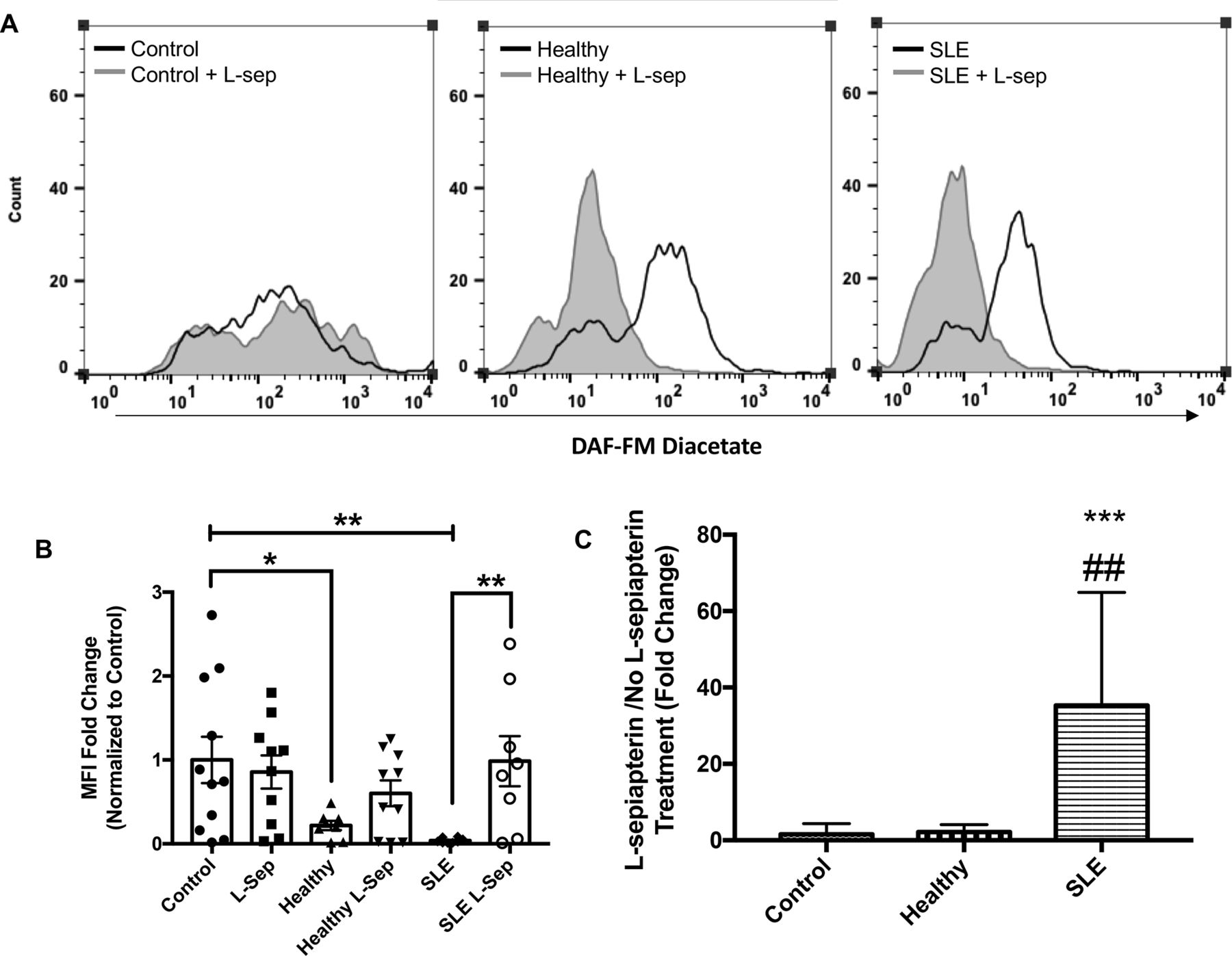
As shown in figure 2A,B, exposure to SLE serum resulted in reduced synthesis of intracellular basal NO as evidenced by reduced DAF-FM diacetate MFI (p<0.01; 23.6-fold) compared with EBM-2 cultured endothelial cells. Moreover, HUVEC exposure to healthy serum caused a reductions in NO compared with EBM-2 but these differences were not as pronounced as changes observed with SLE serum (p<0.05; 4.56-fold).
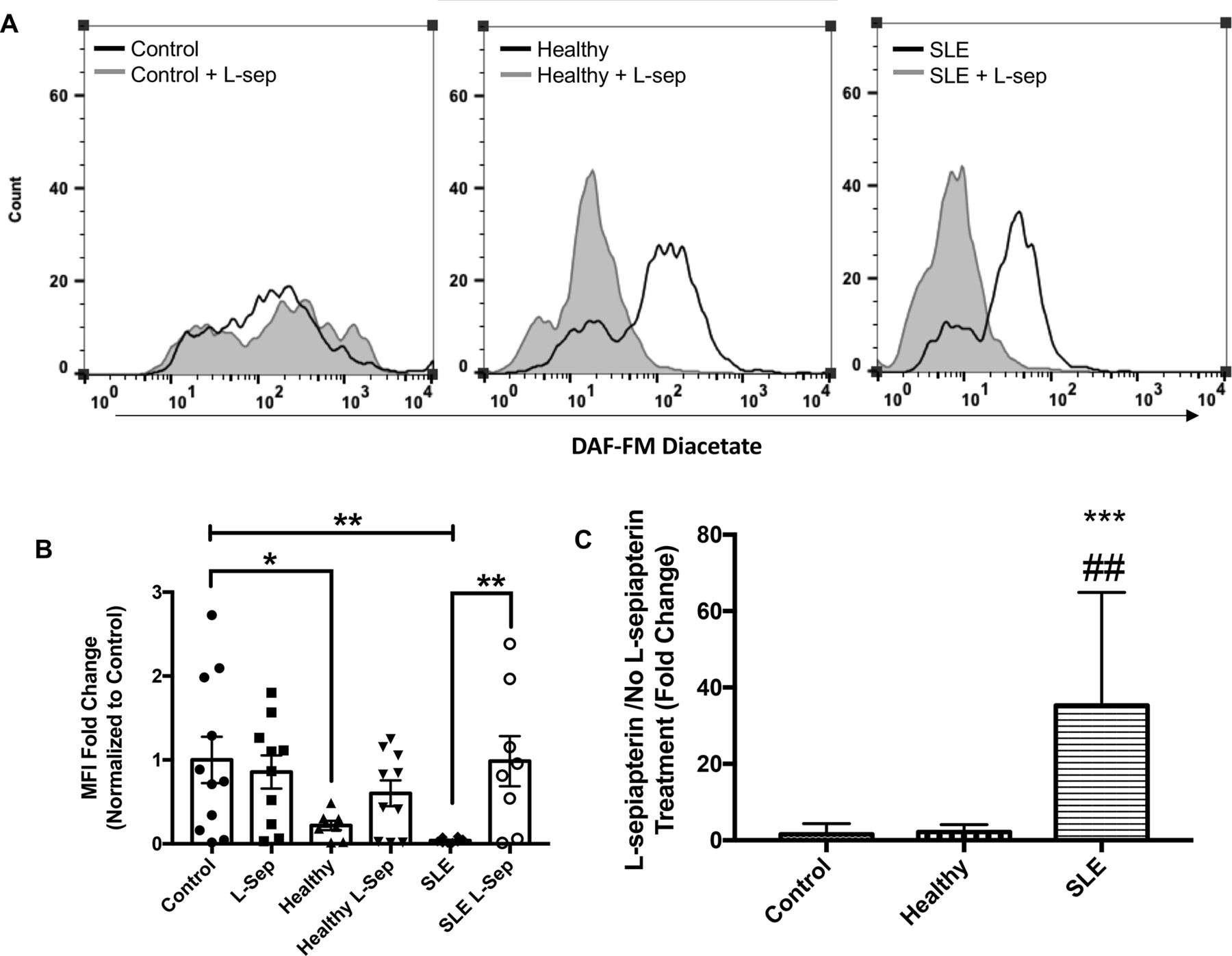
L-sepiapterin (L-sep) restores nitric oxide (NO) production in SLE sera cultured humanumbilical vein endothelial cells (HUVECs). (A) Representative histograms of DAF-FM fluorescence intensity measured in HUVECs cultured in control conditions (grey) or control conditions+L-sepiapterin (black) in the following order endothelial basal medium 2 (EBM-2) buffer (far left), healthy control serum (middle) and SLE serum (far right). (B) A column graph representing the mean fold change of the median fluorescence intensity normalised to buffer controls±SEM. Analysis was conducted using a two-way analysis of variance (ANOVA) with Fisher’s least significance difference post-hoc test. *p<0.05, **p<0.01. (C) A graph representing the mean fold change of L-sep/no L-sep for each serum treatment group. Analysis was conducted using a one-way ANOVA. ***p<0.001, compared with buffer control, ##p<0.01 compared with healthy control.
L-sep restores NO production in SLE serum cultured endothelial cells
Treatment with L-sep, important for tetrahydrobiopterin synthesis (see figure 3), significantly increased DAF-FM MFI, a marker of NO production, in SLE serum cultured HUVECs compared with SLE cultured cells without L-sepiapterin (p<0.01; figure 2B). These changes were not observed in control serum cultured HUVECs. Moreover, we observed that the ratio in MFI-fold change between SLE serum+L-sepiapterin and SLE serum was significantly different from ratios in control and healthy serum culture conditions (p<0.001 and <0.01, respectively; figure 2C).

Postulated impact of SLE on endothelial nitric oxide (NO) synthase activity and endothelial nitric oxide synthase (eNOS) uncoupling. (A) In normal conditions, basal eNOS oxidises L-arginine to L-citrulline and produces NO. However, in the presence of components found in lupus serum, NAPDH oxidase is activated leading to reactive oxygen species and subsequent production of peroxynitrite. (B) Supplementation of low-dose L-sepiapterin in cell cultures raises intracellular BH4 levels leading to improvements in NO production in lupus serum cultured cells.
SLE serum does not alter cellular bioenergetics in endothelial cells compared with healthy serum
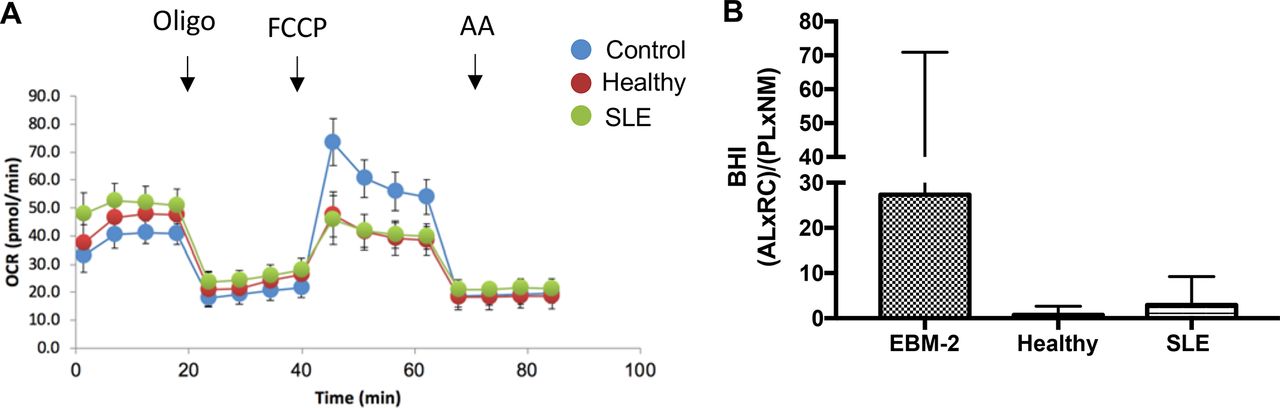
Mitochondrial dysfunction is reflective of changes in mitochondrial membrane potential, a reduction in the ATP level and the inhibition of mitochondrial OCR.38 In addition, excessive ROS contributes to mitochondrial dysfunction as assessed by increased maximal respiration.39 Thus, we sought to determine whether cellular bioenergetics were impacted in response to SLE serum in order to determine the role of mitochondrial oxidative stress in diminished NO production. Figure 4A illustrates the profile of endothelial cells from the three cell culture conditions (EBM-2, healthy serum, SLE serum) after 24 hours. The BHI has previously been shown to be a sensitive measure of oxidative stress and inflammation in various cell types (citation). On calculating the BHI (figure 4B), we found no statistically significant differences in BHI indicating that changes in mitochondrial metabolism do not explain differences in NO production between treatment groups.

Serum does not cause mitochondrial dysfunction. (A) The cellular mitochondrial profile of humanumbilical vein endothelial cells in different cell culture conditions defined by the use of the inhibitors, oligomycin (oligo), FCCP and antimycin A (AntiA). (B) The bioenergetic health index of endothelial cells in buffer (2% fetal bovine serum), healthy control serum (50% v/v), or SLE serum (50% v/v), calculated based on the cellular mitochondrial profile. Analysis was conducted using a one-way analysis of variance and Fisher’s least significance difference post-test. BHI, bioenergetic health index; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone; OCR, oxygen consumption rate.
SLE serum induced neutrophil migration
SLE serum (50% v/v) cultured HUVECs displayed an increased capacity for induction of neutrophil migration as assessed using flow cytometry and confocal microscopy compared with healthy control serum cultured cells (0.50±0.15 vs 1.00±0.54, p<0.01; figure 5A,B). SLE cultured cells also enhanced neutrophil adhesion to the endothelial cell surface compared with cells cultured in EBM-2 media but not healthy control serum. However, these differences were not statistically significant (p=0.07; figure 6A,B). It should be noted that 83% of the SLE patient serum samples used for these experiments were derived from patients who were taking stable doses of prednisone and at least one additional immunosuppressive therapy medication which may have impacted the robustness of neutrophil adherence to the endothelial cell surface under SLE culture conditions.

Lupus serum induces neutrophil chemotaxis. (A) Humanumbilical vein endothelial cells (HUVECs) were stimulated for 6 hours with 50% serum and transwell inserts containing 50 000 calcein AM neutrophils/insert were placed in the well as outlined in the ‘Materials and methods’ section. Images represent calcein am (green) stained neutrophils (first column), bright field +calcein AM stained (second column) of HUVEC cells exposed to endothelial basal medium 2 (EBM-2) (control), Interleukin (IL)-8 (positive control) healthy serum, or SLE serum (B) The graph represents the mean ratio of endothelial cells/neutrophils±SEM n=5 SLE, n=5 SLE +LN, n=5 healthy, n=3 buffer controls. All data were analysed using Kruskal-Wallis analysis of variance and Dunn’s post-test.++p<0.01 compared with healthy control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
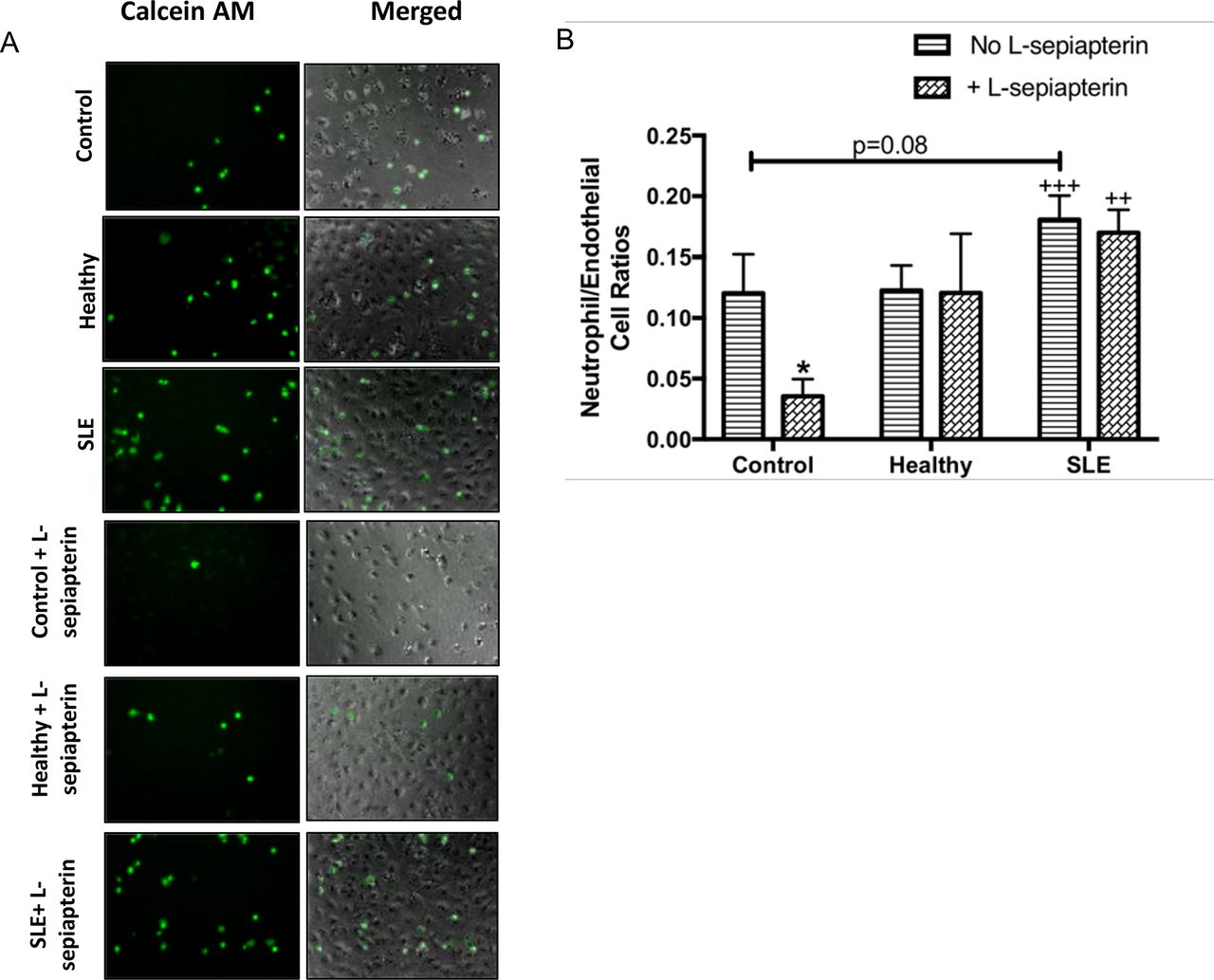
Lupus serum induces neutrophil adhesion to the endothelial cell surface. (A) Humanumbilical vein endothelial cells (HUVECs) were stimulated for 6 hours with 10% serum for 6 hours and stimulated cells were exposed to neutrophils isolated from fresh human blood as outlined in the ‘Materials and methods’ section. images are representative of 6/8 SLE sera and 4/5 control samples. Images represent calcein AM (green) stained neutrophils (first column), bright field +calcein AM stained (second column) of HUVEC cells exposed to endothelial basal medium 2 (EBM-2) (control), healthy serum, or SLE serum in the presence or absence of L-sepiapterin, 40× magnification. (B) Graph represents fluorescence intensity ±SEM. Quantification of neutrophils are reported as means±SEM n=9 SLE, five controls. *p<0.05 compared with EBM-2,++p<0.01 and+++p<0.001 compared with EBM-2 +L-sepiapterin. All data were analysed using two-way analysis of variance and Fisher’s least significant difference post-test.
To determine whether the observed changes in neutrophil adhesion were due to eNOS uncoupling and subsequent declines in NO production, we supplemented cell cultures with L-sep to further understand the role of NO in the observed paradigm. L-sep reversed neutrophil adhesion in cell cultured in EBM-2 media (p<0.05); however, L-sep did not reverse neutrophil adhesion induced by SLE serum (figure 6A,B). These results suggest that other factors beyond NO might be responsible for changes in neutrophil adhesion to the endothelial cell surface.
Discussion
In this study, we observed that SLE serum greatly enhanced eNOS mRNA expression, a phenomenon negatively associated with age. In addition, we observed that SLE serum diminished basal NO in endothelial cells and that L-sep restored levels to normal. While cellular bioenergetics contribute to changes in cell metabolism and signalling pathways, we did not observe differences between the impact of SLE and healthy serum on cellular bioenergetics. Increased levels of neutrophil migration were observed under SLE serum versus healthy serum culture conditions. Moreover, neutrophil adhesion also increased; however, L-sep did not reverse the impacts of SLE on neutrophil adhesion to the endothelial cell surface. Together, these data provide the rationale for the hypothesis that preservation of eNOS function and subsequent NO production may play an important role in protecting against SLE-mediated endothelial dysfunction.
Multiple studies have shown that conventional cardiovascular disease risk factors cause an increase rather than decrease in eNOS expression.40 41 Moreover, while mechanisms involved in SLE serum induced changes in eNOS gene expression are unknown, SLE serum contains large quantities of Crib receptor ligands, including anti-endothelial cell and double-stranded DNA autoantibodies.42 Previous studies demonstrated that FcγRIIB receptor engagement yields disruption in eNOS enzymatic activity and NO production.43 44 Whether these factors trigger activation of pathways that lead to compensatory increases eNOS expression is unknown.
Interestingly, SLE patient age negatively correlated with eNOS mRNA expression. Chronological age is an important non-modifiable cardiovascular risk factor in SLE and non-SLE populations. The relationship between SLE patient age and in vitro endothelial cell eNOS mRNA expression is paradoxical due to conflicting results showing an overall increase in eNOS mRNA expression in SLE. Age is associated with rises in oxidative stress which contributes to progressive declines in endothelium-dependent vasodilation associated with diminished NO bioavailability. Moreover, with age, traditional risk factors including hypertension and dyslipidaemia increase in prevalence.45 We postulate that inflammatory cytokines such as IFN-α, the presence of oxidative stress and the prevalence of conventional cardiovascular disease risk factors could work synergistically to suppress eNOS mRNA expression with age in patients with SLE. However, this hypothesis was not tested in this study.
The role of NO in lupus is multifaceted and oppositional in specific clinical manifestations of the disease. Recent work has suggested that the imbalance between [NO]/[ONOO-] significantly contributes to the development of endothelial dysfunction.46 In SLE, it is postulated that this imbalance stems from a non-resolving inflammatory response42–44 47 that leads to impaired eNOS protein expression and activation, enhanced ROS generation, conversion of BH4 to BH2 and subsequent perpetuating of eNOS uncoupling.48 In the current study, L-sep preserved NO production in response to SLE serum. The preservation of NO production after L-sep treatment may have resulted from increased de novo BH4 synthesis; still, future studies are needed to assess L-sep-mediated preservation of NO. However, a rise in endogenous NO production failed to prevent neutrophil adhesion. These findings contradict the established concept that NO can reduce adhesion molecule expression.49 The reason for this discrepancy is likely multifactorial including the fact that mechanisms mediated by NO are concentration dependent and we did not assess NO production quantitatively but rather qualitatively. Moreover, our studies relied on the use of endogenous rather than exogenous NO previously shown to prevent oxidation of LDL,50 smooth muscle cell migration51 and adhesion molecule expression.52 Still, these studies did not assess the effectiveness of exogenous NO under opposing inflammatory conditions. Our findings are more consistent with in vitro data showing increases in eNOS gene expression coupled with losses in NO production are indicative of dysfunctional eNOS which produces higher levels of superoxide.53 Peroxynitrite, a molecule generated through the interaction of NO and superoxide, oxidises BH4 to BH2 which is not adequate for eNOS enzymatic activity or production of biologically effective concentrations of NO. Thus, oxidation of de novo BH4 could also serve as a potential mechanism of impaired NO bioavailability; however, we did not assess BH4 concentrations in this study. Alternatively, mitochondrial dysfunction may contribute to losses in viable BH4.
Endothelial cells have a very low, mitochondrial content and predominately rely on glycolysis for glucose oxidation and fatty acid oxidation flux.54 As a result, endothelial cells produce lower levels of oxphos-generated ROS and adjust more readily to hypoxic environments.55 Under certain conditions, however, mitochondria become dysfunctional and produce higher levels of ROS. In our studies, the basal OCR increased in response to human serum (both control and SLE). We would postulate that this is expected given the increase in fatty acids and glucose introduced into the cell culture environment. Moreover, we did not observe differences in bioenergetic health indices between human serum groups, which suggests that SLE serum-specific contents do not induce mitochondrial dysfunction and that mitochondrial oxidative stress was not likely the cause of reductions in bioavailable NO in the SLE group.
Our study demonstrated that SLE serum cultured endothelial cells have an increased capacity to induce migration of neutrophils to the endothelial cell surface and bind neutrophils compared with control serum or buffer cultured cells. These findings suggest that factors present in SLE serum may induce adhesion molecule expression, possibly through diminished NO. Previous studies suggest that diminished NO bioavailability further promotes leucocyte adhesion to the endothelial cell surface as NO is known to be a modulator of adhesion and emigration.49 56 Surprisingly, supplementation of cultures with L-sep did not reverse neutrophil adhesion to the endothelial cell surface induced by SLE serum. This suggests that other factors present in SLE serum, including cytokines and chemokines, may promote adhesion molecule expression that cannot be overcome by endogenous NO production in an in vitro cell culture model. Future studies will need to specifically target the NO pathway to differentiate between the role of diminished NO and the inflammatory burden in promoting neutrophil adhesion in SLE serum cell cultures. Moreover, dose–response experiments examining therapeutic concentrations of BH4 in vitro are needed.
This study has several limitations. First, our sample size for these analysis were small; thus, results must be interpreted with caution. Moreover, the clinical profile of healthy controls was incomplete. Thus, we cannot rule out the possibility that they may have had cardiovascular disease risk factors present that attributed to observed alterations in NO production. In addition, we did not examine changes in protein expression or phosphorylation of eNOS in response to healthy control and SLE serum. However, based on our preliminary data, we did observe changes in eNOS dimerisation in response to SLE serum which may account for altered NO production (data not shown). Previous studies assert that these changes rather than changes in mRNA expression are more important contributors to endothelial dysfunction. One caveat to this study is the lack of investigation into the impact of higher concentrations of BH4 on these pathways independent of its effect on eNOS coupling. For instance, use of higher concentrations could have resulted in non-specific superoxide scavenging, which may have impeded our ability to truly assess the isolated role of eNOS coupling in our experiments. Another limitation is our incomplete understanding of specific factors present in SLE serum that may be responsible for the observed effects on endothelial cell function. However, these data provide a proof of concept that SLE serum interferes with endothelial cell function by disrupting eNOS activity and NO bioavailability and that L-sep might be a viable therapeutic option for preventing or restoring the effects of SLE on the endothelium if the dose–response relationship is thoroughly understood.
In summary, endothelial dysfunction is due, in part, to dysfunctional eNOS and diminished NO bioavailability. Lupus serves as an independent risk factor for endothelial dysfunction and contributes to accelerated atherosclerosis.57 Our data provide evidence for dysfunctional eNOS in vitro in the presence of SLE serum.
Acknowledgments
The authors thank Dr. Diane Kamen and Jackie Eudaly for providing and processing SLE patient samples.
References
Footnotes
Contributors JNJB designed and conducted experiments and wrote and edited the manuscript. DPJ conducted experiments and edited the manuscript. RM-H edited the manuscript. JCO wrote and edited the manscript.
Funding The authors are supported by the following: DPJ by National Institute of Diabetes and Digestive and Kidney Diseases T32DK083262 and National Institute of Dental and Craniofacial Research T32DE017551, JNJB by National Institute of Arthritis and Musculoskeletal Diseases 5F31AR06415002, JCO by R01 AR045476, P30 AR072582, and VA Merit Award I01CX001248, JCO and clinical cohort by P60 AR062755, and National Center for Advancing Translational Sciences UL1 TR001450.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval Institutional Review Board at the Medical University of South Carolina.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data, protocols and materials will be made available to researchers upon request and disclosed where restrictions apply.